Article Text

Abstract
Objective: To clarify the effect of interleukin (IL) 18 on cartilage degeneration by studying the profile of IL18 receptor (IL18R) on chondrocytes and the direct effect of IL18 on production of matrix metalloproteinases (MMPs), aggrecanases, and tissue inhibitors of metalloproteinases (TIMPs) in articular chondrocytes.
Methods: Monolayer cultured human articular chondrocytes were isolated from non-arthritic subjects and patients with rheumatoid arthritis or osteoarthritis. Gene expression of IL18, IL18Rα, IL18Rβ, MMPs, and aggrecanases was detected by RT-PCR. Protein levels of IL18Rα were analysed by flow cytometry. Protein levels of IL18, MMPs, and TIMPs were measured by ELISA. Aggrecanase-2 mRNA expression was quantitatively analysed by real time RT-PCR. Protein levels of signalling molecules were assayed by western blotting.
Results: IL18 mRNA was constitutively expressed in chondrocytes, and was enhanced by IL1β stimulation. Flow cytometric analysis showed that IL1β, tumour necrosis factor α, and IL18 up regulated IL18Rα expression levels. The level of IL18Rβ mRNA was much lower than that of IL18Rα, and was slightly up regulated by IL1β. In chondrocytes responding to IL18, IL18 (1–100 ng/ml) slightly increased the production of MMP-1, MMP-3, and MMP-13, which was blocked by NF-κB inhibitor and p38 mitogen activated protein kinase inhibitor. IL18 up regulated mRNA expression of aggrecanase-2, but not aggrecanase-1. IL18 also slightly stimulated TIMP-1 production?through extracellular signal regulated kinase activation.
Conclusion: IL18 induces production of MMPs from chondrocytes in inflammatory arthritis. Although the direct effect of IL18 on chondrocytes may not be pivotal for the induction of cartilage degeneration, IL18 seems to play some part in the degradation of articular cartilage in arthritis.
- DMEM, Dulbecco’s modified Eagle’s medium
- ELISA, enzyme linked immunosorbent assay
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
- IL, interleukin
- IL18R, interleukin 18 receptor
- JNK, c-Jun N-terminal kinase
- MAPK, mitogen activated protein kinase
- MEK, mitogen activated protein kinase/extracellular signal regulated kinase kinase
- MMP, matrix metalloproteinase
- NF-κB, nuclear factor κB
- OA, osteoarthritis
- PGE2, prostaglandin E2
- RA, rheumatoid arthritis
- RT-PCR, reverse transcriptase-polymerase chain reaction
- TIMPs, tissue inhibitors of metalloproteinases
- TNFα, tumour necrosis factor α
- interleukin 18
- articular cartilage
- chondrocytes
- metalloproteinases
- osteoarthritis
Statistics from Altmetric.com
- DMEM, Dulbecco’s modified Eagle’s medium
- ELISA, enzyme linked immunosorbent assay
- GAPDH, glyceraldehyde-3-phosphate dehydrogenase
- IL, interleukin
- IL18R, interleukin 18 receptor
- JNK, c-Jun N-terminal kinase
- MAPK, mitogen activated protein kinase
- MEK, mitogen activated protein kinase/extracellular signal regulated kinase kinase
- MMP, matrix metalloproteinase
- NF-κB, nuclear factor κB
- OA, osteoarthritis
- PGE2, prostaglandin E2
- RA, rheumatoid arthritis
- RT-PCR, reverse transcriptase-polymerase chain reaction
- TIMPs, tissue inhibitors of metalloproteinases
- TNFα, tumour necrosis factor α
The extracellular matrix of articular cartilage is composed primarily of proteoglycans and collagens, which form a network whose integrity provides the mechanical properties required for normal cartilage function. Degradation of extracellular matrix components is often seen as a typical pathological characteristic of rheumatoid arthritis (RA) and osteoarthritis (OA). The degradation of cartilage in vitro and in vivo is believed to be mediated by neutral endopeptidases of the metalloproteinase (MMP) class of enzymes.1,2 Identification of MMP-specific collagen cleavage products in human RA and OA cartilage has established a significant correlation between collagenase activities and tissue destruction during these diseases.3,4 It is well known that MMP activity is controlled by tissue inhibitors of metalloproteinases (TIMPs). An excess of MMPs over TIMPs may contribute to cartilage degradation in OA and RA.4
Interleukin 1β (IL1β) and tumour necrosis factor α (TNFα) have been convincingly shown to promote cartilage degradation by stimulating the production of MMPs.5–7 IL18 has structural similarities to the IL1 family of proteins.8 The receptor complex for IL18, IL18R, consists of two components, an IL18 binding α chain (IL18Rα) and a signalling β chain (IL18Rβ), both of which are required for IL18 signalling.9 Both these IL18R components are members of the IL1R family. Recently, the presence of IL18 was demonstrated in the synovial fluid of patients with RA or OA,10 and a raised level of IL18 was found in canine cartilage with experimental OA.11 Several studies in animal models have shown that IL18 contributes to the destruction of cartilage and bone in the disease process of arthritis.10,12–18 It has also been shown that IL18 stimulates the expression of several genes in normal human articular chondrocytes, including inducible nitric oxide synthase, inducible cyclo-oxygenase, IL6, and stromelysin.14 However, the precise effects of IL18 on the production of collagenases and aggrecanases from chondrocytes remain unknown. Also, the characteristic of IL18R expression in chondrocytes still remains unclear.
Here, we evaluate the expression level of IL18R on chondrocytes and quantitatively analyse the protein levels of two major collagenases (MMP-1 and MMP-13), MMP-3, and TIMP-1 and TIMP-2, as well as mRNA levels of aggrecanase-1 and aggrecanase-2 in cultured chondrocytes after IL18 stimulation. These findings are helpful in furthering our understanding of the direct effect of IL18 on cartilage destruction.
PATIENTS AND METHODS
Materials
A nuclear factor-κB (NF-κB) inhibitor Bay 11-7085, a phosphatidylinositol 3-kinase (PI3K) inhibitor LY294002, a p38 mitogen activated protein kinase (MAPK) inhibitor SB203580, indometacin, and blend collagenase were obtained from Sigma (St Louis, MO, USA); ISOGEN RNA extraction kit from Nippon Gene (Toyama, Japan); recombinant human IL18 from MBL (Nagoya, Japan); a mitogen activated protein kinase/extracellular signal regulated kinase kinase (MEK) inhibitor PD98059 from Wako (Osaka, Japan); c-Jun N-terminal kinase (JNK) inhibitor SP600125 (JNK inhibitor II) from Calbiochem (Darmstadt, Germany); antihuman IL18Rα monoclonal antibody from R&D System (Minneapolis, MN, USA); phycoerythrin conjugated goat F(ab′)2 fragment antimouse IgG (H+L) from Immunotech (Marseille Cedex, France). Ribonuclease inhibitor (porcine liver), random primer (hexa-deoxyribonucleotide mixture), DNA ladder, recombinant Taq DNA polymerase, 10×PCR buffer, 2.5 mM dNTP mixture for PCR from TaKaRa Biomedicals (Shiga, Japan); SuperScript II RNase H reverse transcriptase, 10 mM dNTP Mix, and Zero Blunt TOPO PCR cloning kit from Invitrogen Life technologies (Carlsbad, California, USA); QIAprep spin miniprep kits from QIAGEN Inc (Valencia, CA, USA); LightCycler-FastStart DNA Master SYBR Green I and LightCycler primer sets for human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from Roche Diagnostics GmbH (Mannheim, Germany).
Patients
Fresh articular cartilage specimens were obtained at the time of knee or hip joint replacement surgery from 14 patients with OA (mean age 66 years, range 48–89) and 10 patients with RA (mean age 52 years, range 39–67). All patients met the American College of Rheumatology criteria for RA19 or OA,20 respectively. Normal (macroscopically intact) human cartilage was collected from eight patients (mean age 60 years, range 59–81) with no history of joint disease undergoing joint surgery after traumatic injury. All samples were obtained with informed consent from the patients, and the study protocol was approved by the institutional ethics committee.
Isolation and culture of chondrocytes
Articular cartilage fragments were digested overnight on a horizontal shaker at 37°C in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen Corporation, New York, USA) and 1.0 mg/ml of bacterial collagenase. The cells released by the enzymatic digestion were filtered through a 70 μm nylon filter and collected by centrifugation, and then resuspended and washed four times with collagenase-free medium. The cultures were maintained at 37°C in a 5% CO2 humidified atmosphere in DMEM containing 10% fetal calf serum, 2 mM l-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. Medium was replaced every week. To study the exact expression of IL18 and its receptor, cultured chondrocytes of the second or third passages were starved for 24 hours in DMEM supplemented with 1% fetal calf serum before experiments. To clarify whether or not chondrocyte dedifferentiation affects expression of IL18 and its receptor and the IL18 response, we measured the production of collagen type II and proteoglycan released from cultured chondrocytes by enzyme linked immunosorbent assay (ELISA; type II collagen detection kit: Chondrex, Redmond, WA, USA; proteoglycan detection kit: Biosoure International, Camarillo, CA, USA).
Reverse transcriptase-polymerase chain reaction (RT-PCR) and quantitative real time RT-PCR
Total RNA was extracted from chondrocytes by acid guanidine-phenol-chloroform extraction using ISOGEN. RNA was evaluated spectrophotometrically for quantity and purity. First strand complementary DNA (cDNA) was synthesised from isolated RNA with 0.5 mM dNTP, 10 mM DTT, 15 ng/μl of random primers (hexamers), 2 U/μl of ribonuclease inhibitor, and 10 U/μl of Superscript II reverse transcriptase, and used as templates for PCR. PCR amplification was performed using specific primers (table 1). The constitutively expressed gene encoding GAPDH was used as an internal control in RT-PCR to normalise the amounts of mRNA in each sample. The PCR products were analysed by electrophoresis in 2% agarose gels stained with ethidium bromide, and bands were visualised and photographed under ultraviolet excitation.
Sequences of PCR primers, length of PCR product, optimal annealing temperature, and sequences accession number from the NCBI Entrez search system
Quantitative real time RT-PCR was performed with a spectrofluorometric thermal cycler (LightCycler, Roche Diagnostics). For each run, a standard curve was generated from purified DNA ranging from 105 to 10 copies of the cDNA of interest. Specificity of the expected products was demonstrated by melting curves analysis. To standardise mRNA levels, we amplified GAPDH as internal control by using a commercial primer set according to the manufacturer’s protocol. Normalised gene expression was calculated as the ratio between the copy number of the gene of interest and that of GAPDH cDNA.
ELISA
The levels of total MMP-1, total MMP-3, proMMP-13, TIMP-1, and TIMP-2 in supernatants were evaluated with commercial ELISA kits according to the instructions of the manufacturer. The ELISA kits for total MMP-1, proMMP-13, and TIMP-2 were purchased from Amersham Biosciences (Little Chalfont, Buckinghamshire, UK); for total MMP-3 from R&D System (Minneapolis, MN, USA); and for TIMP-1 from Daiichi Fine Chemical (Toyama, Japan). The sensitivities of the assays were 1.7 ng/ml for MMP-1, 9 pg/ml for MMP-3, 0.032 ng/ml for MMP-13, and 3.0 ng/ml for TIMP-2. The level of IL18 in supernatants was assayed with the ELISA kit from MBL (Nagoya, Japan) with a sensitivity of 12.5 pg/ml.
Western blotting analysis
The protein concentrations of the whole cell lysates were determined by a Bradford assay (Bio-Rad). Equal amounts of protein were loaded into an sodium dodecyl sulphate-polyacrylamide gel (10% acrylamide), resolved by electrophoresis, and transferred to polyvinylidene difluoride membranes. After blocking with 5% skimmed milk in phosphate buffered saline/0.1% Tween-20, the protein expression was determined using specific antibodies purchased from the following sources: goat anti-phospho-Erk1/2 (p-Erk1/2), rabbit anti-Erk1/2, rabbit anti-phospho-p38 (p-p38) and rabbit anti-p38 (Cell Signalling Technology, Beverly, MA), goat anti-phosphorylated IκB (p-IκB) and mouse monoclonal antibody to actin (Santa Cruz Biotechnology, Santa Cruz, CA). The blots were developed using a horseradish peroxidase conjugated secondary antibody and an enhanced chemiluminescent system (Amersham Pharmacia Biotech, Piscataway, NJ).
Statistical analysis
All experiments were repeated independently, at least three times. Data were expressed as a ratio of experimental to control values (mean (SD)) for the chondrocytes responsive to IL18. Comparisons between groups were made by Student’s t test and analysis of variance. Values of p<0.05 were considered significant.
RESULTS
Gene expression and production of IL18 in chondrocytes
During the continuous culture of chondrocytes (up to five passages), no significant differences in the production of type II collagen and proteoglycan were seen (fig 1), suggesting that chondrocyte dedifferentiation did not affect the results of this study.
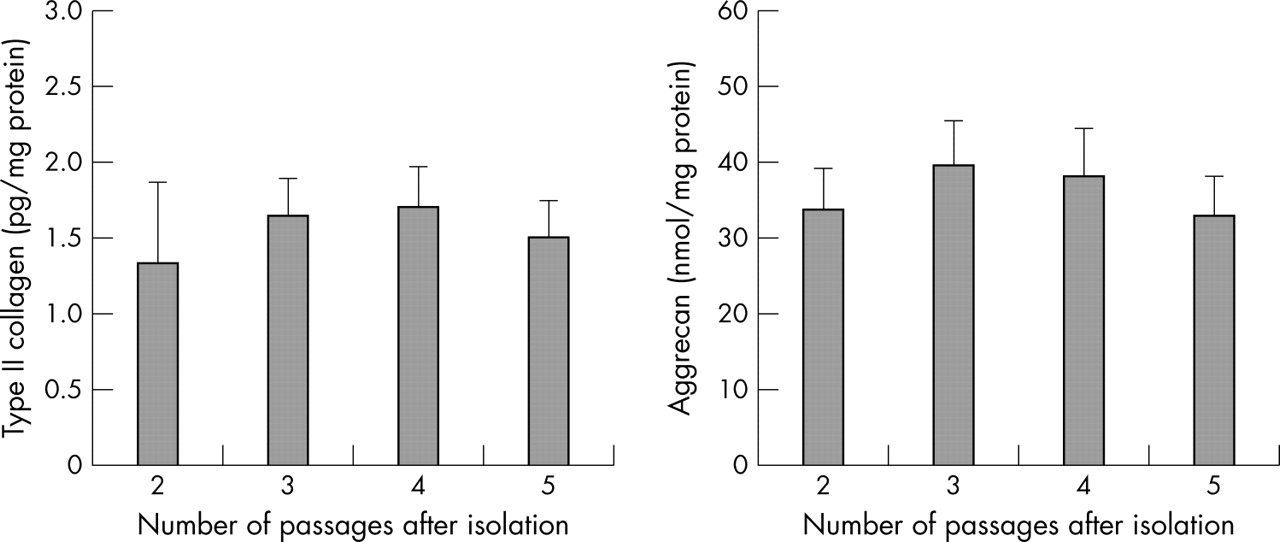
Production of type II collagen and proteoglycan during the continuous culture of chondrocytes. The levels of production of type II collagen and proteoglycan released by cultured chondrocytes were analysed by an ELISA.
IL18 mRNA was constitutively expressed in cultured chondrocytes from normal cartilage, OA cartilage, or RA cartilage (fig 2A), and was up regulated by 10 ng/ml IL1β (fig 2B). However, the baseline level of soluble IL18 protein in culture supernatants was undetectable by ELISA (the cut off point is 25.6 pg/ml). Furthermore, it was still undetectable after IL1β (1–25 ng/ml) or TNFα (2∼50 ng/ml) or prostaglandin E2 (PGE2, 1∼25 μM) stimulation for 48 hours.

Expression of mRNA for IL18, IL18Rα, and IL18Rβ by cultured chondrocytes. The chondrocytes were isolated from normal human joint cartilage, OA and RA cartilage, and mRNA expression was detected by RT-PCR. PCR products were stained with ethidium bromide. Results are representative of individual experiments (A). Effect of IL1β (10 ng/ml) on the expression of IL18 (B), IL18Rα (C), and IL18Rβ (D) were analysed by real time PCR.
Expression of IL18R in chondrocytes
Table 2 shows the profile of the cultured chondrocytes expressing IL18R. IL18Rα was not constitutively expressed in chondrocytes isolated from any of the samples, expression was found in only 4/8 (50%) samples of normal cartilage, 9/14 (64%) samples of OA cartilage, and 6/10 (60%) samples of RA cartilage. After IL1β stimulation, the expression level of IL18Rα was significantly up regulated in all cases (fig 2C). The constitutive expression level of IL18Rβ was barely detectable after amplification for 35 cycles by RT-PCR in all the samples. We therefore amplified the PCR product for another 20 cycles using nested primers after an initial amplification for 25 cycles. As a result, the nested PCR showed the expression of IL18Rβ in all cases. IL1β up regulated the expression level of IL18Rβ to some extent (fig 2D). Figure 2A shows representative data of IL18R expression.
Profile of the cultured chondrocytes expressing IL18R or responding to IL18
The changes in IL18Rα expression level after stimulation were also quantitatively analysed by flow cytometry. After 12 hours’ stimulation, IL1β as well as TNFα markedly increased the expression levels of IL18Rα. IL18 also increased the expression level (fig 3).

Flow cytometric analysis of IL18Rα expression on the surface of cultured OA chondrocytes. (A) Representative dot plots of cells incubated with or without IL1β (5 ng/ml), TNFα (10 ng/ml), or IL18 (10 ng/ml) for 12 hours. (B) Changes in mean fluorescence intensity (ΔMFI). **p<0.01 v control.
Up regulation of mRNA levels of MMPs and aggrecanase-2 by IL18
In most cases, IL18 failed to regulate MMP production in cultured chondrocytes. In those chondrocytes responding to IL18, up regulation of mRNA expression levels of MMP-1, MMP-3, MMP-13, and aggrecanase-2 occurred from 1 to 48 hours after stimulation (fig 4A). We also confirmed the enhanced expression level of aggrecanase-2 by quantitative real time RT-PCR (fig 4B).

Time course of IL18 effects on gene expression of MMP-1, MMP-3, MMP-13, and aggrecanase-2 in OA chondrocytes. (A) Representative results from chondrocytes that responded to IL18 by RT-PCR. (B) Expression level of aggrecanase-2 analysed by quantitative real time RT-PCR. The details are described in “Patients and methods”. The copy number of aggrecanase-2 mRNA was standardised by the copy number of GAPDH mRNA. Compared with the control (without IL18): *p<0.05, **p<0.01.
The expression level of aggrecanase-1, normally very low in vivo,21 was also found to be much lower than that of aggrecanase-2 in our cultured articular chondrocytes, and it was not markedly up regulated by IL18 in those chondrocytes which produced more MMPs after IL18 stimulation.
Enhancement of protein levels of MMPs by IL18
Only some of the samples (2/8 (25%) normal, 5/14 (36%) OA, and 3/10 (30%) RA) of chondrocytes responded to IL18 stimulation (table 2). We found no differences between the clinical data of IL18 responsive cartilage samples and non-responsive samples. In chondrocytes that responded to IL18, IL18 enhanced MMP-1, MMP-3, and MMP-13 production for normal, OA, and RA chondrocytes in a dose dependent manner. There was no significant difference in the magnitude of the enhancing effect among the chondrocytes isolated from the normal joint, or from OA or RA cartilage. The maximal increase for these MMPs was about twofold greater than that of the control after 100 ng/ml IL18 stimulation (fig 5), whereas 1 ng/ml IL1β enhanced the mean (SD) production of MMP-1 to 16.1 (11.4), MMP-13 to 23.3 (9.1), and MMP-3 to 14.1 (6.5) fold greater than that of the control in all cases. Although IL1β up regulated the expression levels of IL18R, we failed to demonstrate a synergistic effect on MMP production when 1 ng/ml IL1β was combined with 1–100 ng/ml IL18.
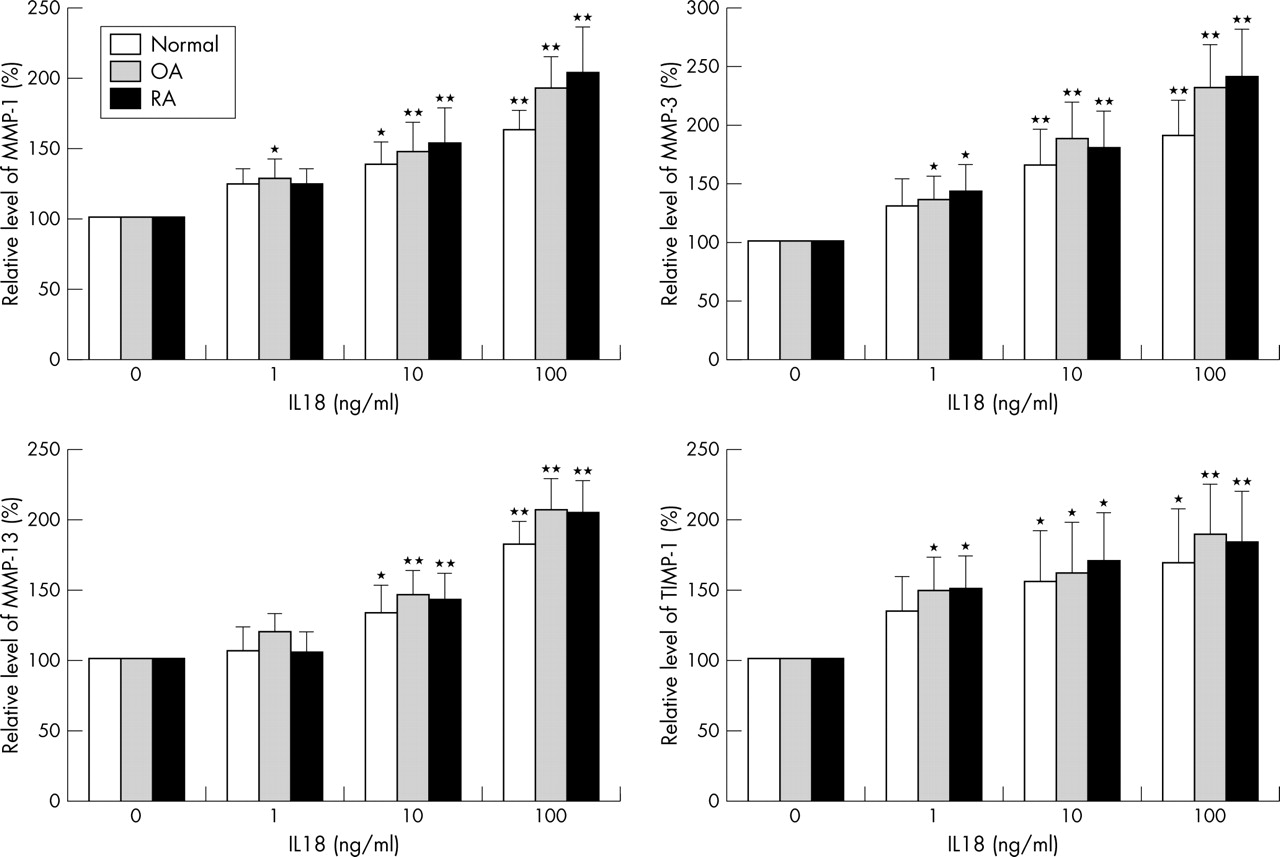
Dose-response of IL18 effects on MMP-1, MMP-3, MMP-13, and TIMP-1 production in chondrocytes. After a 48 hour incubation with indicated concentrations of IL18, the supernatants were harvested. The levels of MMPs and TIMP-1 in the supernatants were assayed by ELISA. Here only the results from the chondrocytes responsive to IL18 were shown. Compared with the control (0 ng/ml IL18): *p<0.05; **p<0.01.
Enhancement of TIMP-1 production by IL18
In those chondrocytes, which produced more MMPs after IL18 stimulation, IL18 also increased the TIMP-1 production in a dose dependent manner (fig 5), but did not show any effect on TIMP-2 production. IL1β had no significant effect on TIMP-1 and TIMP-2 production from chondrocytes. The baseline level of TIMP-2 in supernatants of cultured chondrocytes was much lower than that of TIMP-1.
Signal transduction pathways involved in the effects of IL18
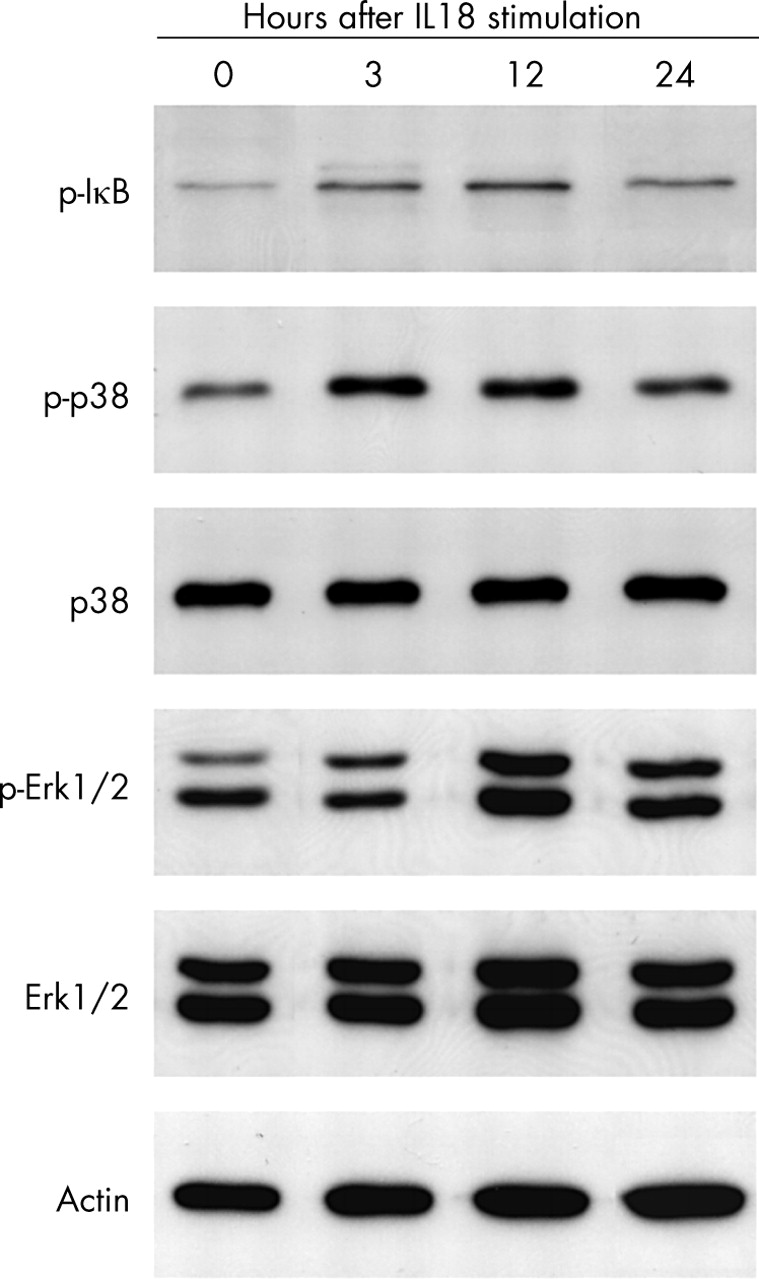
The cultured chondrocytes were pretreated with a specific inhibitor of NF-κB (20 μM Bay 11-7085), PI3K (10 μM LY294002), p38 MAPK (10 μM SB203580), MEK (an upstream enzyme of Erk1/2, 30 μM PD98059), or JNK (20 μM SP600125) 30 minutes before the addition of 100 ng/ml IL18. As a result, the NF-κB inhibitor or p38 MAPK inhibitor suppressed the IL18 enhanced production of MMP-1, MMP-3, and MMP-13, while the MEK inhibitor reduced TIMP-1 production (fig 6). The activation of NF-κB, p38 MAPK, and Erk1/2 induced by IL18 was confirmed by increased levels of phosphorylated IκB, phosphorylated p38 MAPK, and phosphorylated Erk1/2 (fig 7).

Effect of specific inhibitors on IL18 enhanced production of MMP-1, MMP-3, MMP-13, and TIMP-1 in OA chondrocytes. In IL18 responsive chondrocytes, specific inhibitor of NF-κB (20 μM Bay11-7085), PI3K (10 μM LY294002), p38 MAPK (10 μM SB203580), MEK (30 μM PD98059), JNK (20 μM SP600125), inducible nitric oxide synthase (100 μM 1400W), or cyclo-oxygenase (1 μM indometacin) was added to the culture medium 30 minutes before the addition of 100 ng/ml IL18. Compared with the control group (only IL18): *p<0.05; **p<0.01.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Phosphorylation of IκB, p38 MAPK, and Erk1/2 induced by IL18 in responsive OA chondrocytes. Chondrocyte lysates were prepared at the indicated times after stimulation with 100 ng/ml IL18. Whole cell lysates were subjected to western blotting with specific antibodies.
Nitric oxide or PGE2 does not mediate the effects of IL18
It has been reported that IL18 up regulated the gene expression of inducible nitric oxide synthase and inducible cyclo-oxygenase in chondrocytes,14 leading us to apply specific inhibitors of these enzymes to study whether nitric oxide or PGE2 mediates the effects of IL18 on chondrocytes. Neither the nitric oxide synthase inhibitor (100 μM 1400W) nor the cyclo-oxygenase inhibitor (1 μM indometacin) affected the IL18 enhanced production of MMP-1, MMP-3, MMP-13, or TIMP-1 (fig 6).
DISCUSSION
IL18 is a unique cytokine with a wide spectrum of biological actions. We found constitutive expression of IL18 and up regulated expression level of IL18 by IL1β in cultured articular chondrocytes, which is consistent with a previous report.14 However, we found that the level of IL18 in the supernatants of monolayer cultured chondrocytes was too low to be detected by ELISA, even after stimulation with IL1β, TNFα, or PGE2 for 48 hours, which suggests that cartilage releases little IL18 into the synovial fluid in inflammatory arthritis.
In the present study about 50–60% of the samples of chondrocytes constitutively express the IL18Rα chain. Although IL1β or TNFα, key proinflammatory cytokines in synovitis, significantly up regulated the expression level of IL18Rα, there was no significant difference in the number of cases with constitutive IL18Rα expression in chondrocytes from arthritic and non-arthritic groups. The expression level of IL18Rβ in chondrocytes was so low that nested RT-PCR had to be carried out to show its expression. Total RA synovial cells containing T cells showed a strong expression of both IL18Rα and IL18Rβ mRNA.18,22 From the point of the expression level of IL18R, the T cells or macrophages which invaded the synovium or which were in synovial fluid may be the pivotal target mediating the effects of IL18 in RA or OA.
Because IL18 is structurally related to IL1, we compared their effects on MMP-1, MMP-3, and MMP-13 production in chondrocytes. Our data indicated that chondrocytes from only about 30% of the cases were responsive to IL18, and that 100 ng/ml IL18 has only a modest effect on these MMPs, increasing production by about twofold. IL1β showed a much more potent effect on the production of MMPs than IL18. It may be that IL18, compared with IL1β, is too weak to induce MMPs from chondrocytes. Although there was a tendency for IL18 responsive chondrocytes to have higher levels of MMPs than non-responsive ones, no statistically significant correlation was seen. Further studies are needed to analyse the correlation of IL18 response with the expression of MMPs in chondrocytes. In our study we also found that IL18 up regulated the aggrecanase-2 mRNA expression level in OA chondrocytes and slightly increased TIMP-1 production. Based on these data, it is hard to say whether the dominant effect of IL18 on cartilage is destructive or protective in arthritis.23–25
There are four possible routes for the signalling pathways of IL18 (namely, NF-κB, PI3K, p38 MAPK, extracellular signal regulated kinase p42/p44 (Erk1/2) pathways) in other cell types.16,26–28 We found that the enhancement of MMP-1, MMP-3, and MMP-13 depended on the activation of the NF-κB and p38 MAPK pathways, whereas the increased production of TIMP-1 depended on the Erk1/2 pathway. Although IL1β also activates the Erk1/2 pathway,29,30 it has little effect on TIMP-1 production in the present study. This phenomenon can be explained by the fact that activation of p38 MAPK is thought to be a suppressor of TIMP-1 production,31 and that IL1β is an extremely potent inducer of p38 MAPK activation in chondrocytes. In addition, Erk1/2 activation by IL1β in articular chondrocytes is transient: activation is detectable within 5 minutes after IL1 stimulation and has decreased to baseline within 60 minutes.32
In the current study we used PD98059 for MEK inhibitor and SP600125 for JNK inhibitor to analyse the signal pathway of IL18 induced MMP expression in articular chondrocytes. Neither inhibitor prevented IL18 mediated expression of MMPs in articular chondrocytes. Thus, we concluded that MEK and JNK might not be the main pathway for the IL18 effects. Because both inhibitors influence the activity of activator protein-1, these inhibitors might affect the IL18 induced expressions of MMPs. However, in our study, we found no significant differences between PD98059 treated and SP600125 treated groups. Further studies are needed to clarify the exact signalling pathway of IL18.
In conclusion, although cartilage is not the most important source of raised IL18 in synovitis, chondrocytes express IL18 mRNA, which is slightly up regulated by IL1β; IL18 slightly induces MMP-1, MMP-3, MMP-3, and aggrecanase-2 through activation of the NF-κB and p38 MAPK pathways, as well as TIMP-1 through activation of the Erk1/2 pathway in chondrocytes from a small proportion of RA or OA samples. Although the direct effect of IL18 on chondrocytes may not be pivotal for the induction of cartilage degeneration, IL18 may, at least in part, be involved in the articular cartilage degradation in arthritis.