Article Text

Abstract
Objectives Janus kinase inhibitors (JAKi) have been approved for use in various immune-mediated inflammatory diseases. With five agents licensed, it was timely to summarise the current understanding of JAKi use based on a systematic literature review (SLR) on efficacy and safety.
Methods Existing data were evaluated by a steering committee and subsequently reviewed by a 29 person expert committee leading to the formulation of a consensus statement that may assist the clinicians, patients and other stakeholders once the decision is made to commence a JAKi. The committee included patients, rheumatologists, a gastroenterologist, a haematologist, a dermatologist, an infectious disease specialist and a health professional. The SLR informed the Task Force on controlled and open clinical trials, registry data, phase 4 trials and meta-analyses. In addition, approval of new compounds by, and warnings from regulators that were issued after the end of the SLR search date were taken into consideration.
Results The Task Force agreed on and developed four general principles and a total of 26 points for consideration which were grouped into six areas addressing indications, treatment dose and comedication, contraindications, pretreatment screening and risks, laboratory and clinical follow-up examinations, and adverse events. Levels of evidence and strengths of recommendations were determined based on the SLR and levels of agreement were voted on for every point, reaching a range between 8.8 and 9.9 on a 10-point scale.
Conclusion The consensus provides an assessment of evidence for efficacy and safety of an important therapeutic class with guidance on issues of practical management.
- Inflammation
- Therapeutics
- Autoimmune Diseases
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Introduction
The therapeutic options for patients with immune-mediated inflammatory diseases (IMIDs), such as rheumatoid arthritis (RA), psoriatic arthritis (PsA), axial spondyloarthritis/ankylosing spondylitis (AxSpA/AS), systemic lupus erythematosus (SLE), psoriasis (PsO), atopic dermatitis (AD), Crohn’s disease (CD), ulcerative colitis (UC) and others, have significantly improved over the past two decades. This results primarily from the introduction of several novel medications, in particular biological (b) disease-modifying antirheumatic drugs (DMARDs), as reflected in recent management recommendations.1–6 Improved strategic utilisation of drugs has similarly impacted positively on outcomes.
Among all therapies developed for IMIDs over the last two decades, only tumor necrosis factor (TNF)-inhibitors exhibit a very broad efficacy across many diseases: RA, PsA, axSpA, juvenile idiopathic arthritis, PsO, CD, UC and uveitis.7 Even though targeting just a single cytokine, no other treatment modality has yet been approved for such a broad list of indications, suggesting that TNF is pathogenetically involved across a diverse range of IMIDs. All other biological agents are licensed for fewer indications. This will likely change with the advent of Janus kinase (JAK)-inhibitors (JAKi), a new class of targeted synthetic DMARDs (tsDMARDs) that interfere with signal transduction pathways of a variety of cytokines and thereby have the potential to mediate immune modulatory benefits across a broad range of pathologies and their clinical phenotypes.
bDMARDs are usually monoclonal antibodies or receptor constructs that target a specific soluble or cell surface molecule, either a cytokine, a cytokine receptor or another cell membrane antigen. They either prevent interaction of the specific ligand with its cognate receptor, destroy a specific cell population, such as B-cells, or inhibit cross talk between particular cell populations. They have to be administered parenterally since they are proteins. They also do not enter the cell but mediate their respective modes of action outside the cell or via the cell surface.
The pathways that mediate cytokine receptor signal transduction have been elucidated in recent years providing novel and rational targets for drug development to modify cytokine effector function. Synthetic chemical agents that interfere with these pathways have been developed for various indications.8–10 Among them, the JAKi represent a series of intracellularly active drugs, some of which have been approved for the treatment of several IMIDs. Five JAKi, tofacitinib, baricitinib, peficitinib, upadacitinib and filgotinib, are currently approved for therapeutic use in one or more IMIDs in a number of geographical regions.
Our experience with bDMARDs spans two decades across many diseases with thousands of patient years of experience including in registries in many countries. In contrast, data from registries are quite limited for JAKi and some have only recently been approved by several regulatory authorities; safety data for more than 10 years are derived mainly from long term extensions of randomised clinical trials.11 12 Therefore, it was deemed important to develop an evidence-based consensus statement that focuses on practical issues in the use of JAKi.
Scope and purpose
Recommendations for the management of individual IMIDs focus primarily on therapeutic strategies and the general use of individual or groups of agents. While quite comprehensive, they usually address a particular disease and general issues, only rarely accommodating the various, often complex aspects related to the general application of an individual drug or a specific mode of action. Therefore, consensus statements on the more comprehensive use of specific agents or classes of drugs have also been developed.13–16 These provide more detailed information on efficacy and safety of a class of drugs than in the traditional broad management recommendations. Such ‘points to consider’ can provide prescribers, like specialists in specific disease areas, and patients (especially when information is available for laypersons), with an expert opinion on appropriate use of a new drug and its place in treatment algorithms. When a drug is approved for more than one indication, a specific consensus statement can be used across specialties. Thus, the target of the present consensus statement comprises rheumatologists, dermatologists, gastroenterologists, other health professionals involved in these areas, patients with these respective diseases, but also hospital managers and representatives of regulators and social security agencies.
These points to consider are not meant to suggest a preferential use of JAKi for any particular disease but rather to provide evidence-based information in conjunction with expert opinion once an agent of this class has been considered for the treatment of a patient with a specific disease for which the drug is indicated. A research agenda will complement these points to drive momentum to search for more evidence where this is insufficient or lacking. Before addressing the methodology related to this document, we will briefly allude to the mode of action and other pharmacological aspects of this class of drugs.
Mode of action
JAKs are non-receptor tyrosine kinases associated with the cytoplasmic domain of type I and II cytokine receptors which are activated when these are engaged by their cognate ligands; once phosphorylated, they phosphorylate signal transducers and activators of transcription (STATs) which then induce gene activation.17 JAKi reversibly inhibit kinase signalling for varying periods of the dosing cycle. They are oral small molecules that act intracellularly and prevent the phosphorylation of JAKs. Many cytokines, such as interleukin (IL)-2, 6, 12, 15 and 23 as well as interferons use the JAK-STAT pathways, while others, such as IL-1, IL-17 and TNF, do not (figure 1). In addition, haematopoietic growth factor receptors, such as those for erythropoietin (EPO), thrombopoietin and granulocyte-macrophage colony-stimulating factor, use the JAK-STAT pathway (figure 1). Within the cell usually different JAK molecules are associated with each of these receptor chains, acting in tandem as heterodimers, such as JAK1 and JAK2, JAK1 and JAK3 or JAK1 and TYK2. Only in the case of haematopoietic growth factor receptors both chains carry JAK2. Thus, JAK enzymes - JAK1, 2, 3 and TYK2 - function as dimers and once activated phosphorylate STATs, which subsequently induce gene transcription.

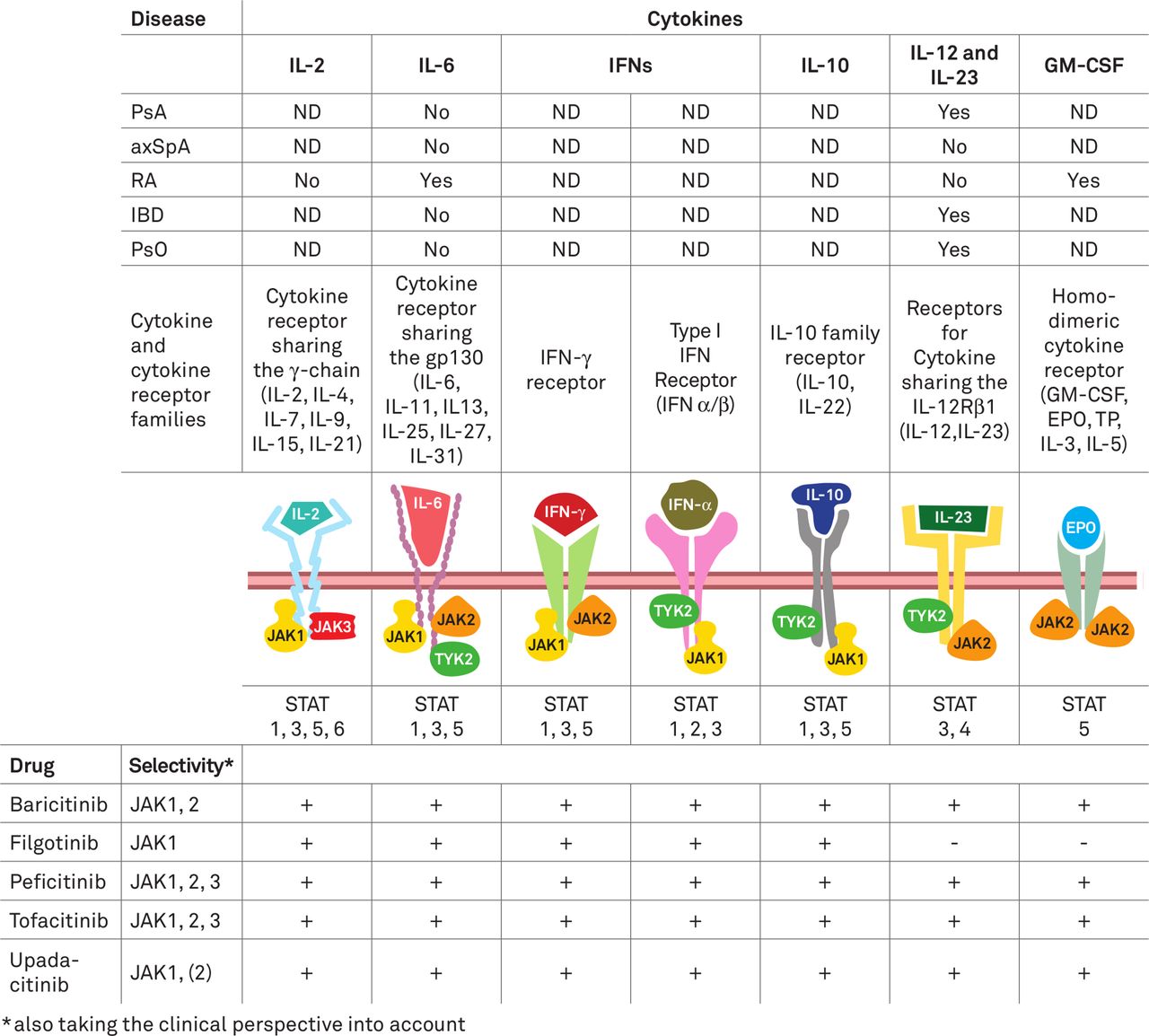{kind=link}
Depiction of cytokines that activate and drugs that target Janus kinases (JAKs) presumably involved in the pathogenesis of immune-mediated inflammatory diseases (IMIDs). Top: efficacy of agents targeting specific JAK-inducing cytokines in different IMIDs. Centre: cytokines and respective receptors that trigger JAKs, types of JAKs activated and type of STATs (signal transducers and activators of transcription) activated by the respective JAKs. Bottom: JAK-inhibitors which are currently approved for IMIDs and their overall (including clinically derived) selectivity and presumed interference (+ or -) with certain cytokine pathways. axSpA, axial spondyloarthritis; EPO, erythropoietin; GM-CSF, granulocyte-monocyte colony stimulating factor; IBD, inflammatory bowel diseases; IFN, interferon; IL, interleukin; ND, not done; PsA, psoriatic arthritis; PsO, psoriasis; R, receptor; RA, rheumatoid arthritis; TP, thrombopoietin.19 40 146 147
The selectivity of JAKs can be determined by using purified enzyme systems and a variety of cellular models.18 19 Varying approaches may lead to differing results with respect to perceived selectivity of JAKs, and selectivity is dose-dependent, since at higher doses the compounds lose selectivity.19 20 The in vivo selectivity may differ further so that in vivo markers may also be helpful. Reduction of inflammation usually produces an increase in haemoglobin, as exemplified by the rapid normalisation of anaemia in patients receiving monoclonal antibodies to the IL-6 receptor.21 Since EPO signals through JAK2 homodimers, failing to see an increase in haemoglobin in patients with anaemia of chronic disease who experience clinical improvement on JAKi therapy suggests an important degree of JAK2 inhibition. Of note, failure to increase haemoglobin is not necessarily linked to fatigue and rarely a reason to stop a JAKi. Current views on the selectivity of JAKi, taking all aspects including clinical ones (such as effects on haemoglobin levels) into consideration are provided in figure 1. Of note, the totality of in vivo downstream effects of JAKi is still insufficiently understood, especially in specific disease settings, and an important matter for further research activities.
Given that individual JAKs and STATs can be activated by more than one cytokine, upregulation and activation of a single STAT pathway does not implicate any one particular cytokine in a response and as such our understanding of the hierarchical contribution of distinct STATs to effector pathways remains conjectural. Nevertheless, success and failure of therapeutic trials of drugs of known selectivity enable some insights into pathogenesis (figure 1). For example, both IL-6 and IL-23 receptors (R) signal via JAKs; since IL-6R inhibition does not appear efficacious in PsA or PsO, while IL-12 and IL-23 inhibition is,22–24 this infers that beneficial effects of JAKi may arise by inhibiting IL-23 rather than IL-6 signalling. In contrast, IL-6R antibodies, but not anti-IL-12/23 antibodies,25 are efficacious in RA and, therefore, JAKi may be assumed to convey efficacy by blocking IL-6 rather than IL-12 or 23 signal transduction. Moreover, neither IL-12, IL-23 nor IL-6R inhibition are efficacious in AS,26 27 while JAKi appear to be28; consequently, this effect cannot be explained by interference with IL-6, IL-12 or IL-23 signal transduction, but rather by inhibition of signal transduction of other cytokines captured by JAKi (figure 1). However, also inhibition of type I (or type II) interferon signal transduction may play a role.29 30 Similar deliberations may be made for inflammatory bowel disease, where IL-6R inhibition is not, or only weakly efficacious,31 while IL-12/23 blockade is efficacious,32 and for PsO.33 On the other hand, while pan JAKi is apparently efficacious in UC but not in CD,34 35 more JAK1 selective inhibitors (filgotinib, upadacitinib) showed promising results in CD,36 37 implying that differences in the pathogeneses of these two inflammatory bowel diseases manifest in subtle but functionally important variations in the relative contribution of these signalling pathways.38 Finally while TNF does not activate the JAK-STAT pathway directly, it might do so indirectly via induction of other cytokines, such as IL-6 or type I interferons.39 This adds further complexity to our understanding of the pathogenesis of IMIDs.
Thus, JAKi via its mode of action across signal transduction of multiple cytokines is efficacious across a range of IMIDs. By corollary, this effect has potential safety repercussions (see below). Clinical experience with JAKi will likely provide innovative insights to rewrite our understanding of IMIDs.
Among the JAKi currently approved or under study for IMIDs, current information on enzyme assays, cellular assays and in vivo data (see above note on laboratory test results regarding JAK2 inhibition, especially anaemia) suggest that at clinically used doses tofacitinib is preferentially a JAK 1, 3 and 2 inhibitor; baricitinib is primarily a JAK 1 and 2 inhibitor; peficitinib is an inhibitor of JAK3 over JAK 1, 2 and TYK2; upadacitinib is a JAK1 inhibitor with effects on JAK2, and filgotinib is primarily a JAK 1 inhibitor (figure 1).40 As mentioned above, the preferential selectivity is dose-dependent and decreases with increasing doses as their common mechanism is to prevent ATP-mediated protein tyrosine kinase phosphorylation (although a specific TYK2 selective inhibitor is also under development that inhibits signal transduction by stabilising the pseudokinase domain of the protein).41
Methods
The expert committee adhered to the EULAR standard operating procedures for the development of recommendations.42 A steering committee comprising 15 members and an expanded Task Force consisting of 14 additional individuals invited based on their expertise and availability and including two patient research partners (MdW, MV) and a health professional (MS-M) as well as a dermatologist (W-HB), a gastroenterologist (MT), a haematologist/haemostaseologist (KG), an infectious disease specialist (KLW) and a fellow who performed the systematic literature review (SLR) (AK), evaluated the available data. The clinicians were all experienced in the treatment of chronic inflammatory diseases, had participated in clinical trials of JAKi and/or bDMARDs, and several had long-standing experience in patient outcomes research and prior consensus statement development. The patients and health professionals all had experience in consensus activities. There was a broad global representation from European countries, Asia, Australia, Latin America and North America. All task force members declared their potential conflicts of interest and had ongoing opportunity to declare if they felt conflicted throughout the process.
Drugs that had not yet undergone regulatory assessment or formal approval but for which evidence from clinical trials was available, could be considered in the recommendations to anticipate potential future uptake in clinical practice, with all respective caveats that may emerge during the approval process. Indeed, during the time of writing or revising the manuscript (and thus after the face-to-face meetings), two drugs, upadacitnib and most recently filgotinib, were approved (at least in some regions), confirming the validity of the conclusions drawn on these agents in the course of the process developing the consensus statement.
The steering committee and the fellow (AK) initially discussed the research questions for the SLR which was then performed accordingly by searching the totality of the respective clinical trial literature until end of December 2018 in Medline, Embase, Cochrane and 2018 EULAR and ACR abstracts. The details of the SLR are published separately.43 Cochrane risk of bias tool was used. The SLR addressed RA, PsA, PsO, AS, systemic lupus erythematosus (SLE), UC, CD, alopecia areata (AA)/alopecia universalis, and atopic dermatitis (AD).
The results of the SLR were first presented to the steering group which developed a list of proposed recommendations and/or topics to be addressed by the whole task force. The SLR and the list prepared by the steering group were then presented to the task force which met end of March 2019. Efficacy aspects were discussed by the whole task force with input from experts in respective fields. The Task Force was split into four breakout groups. One group addressed screening, the second monitoring, the third contraindications and the fourth adverse effects. Representatives of each breakout group reported the results of the deliberations and presented proposals for the wording of individual points to the whole task force which discussed them in detail before voting took place.
For a general principle or point of consideration to be accepted for the final document without further change, a majority of 75% of the votes was required in the first ballot. If this result was not achieved, the respective text was amended and subjected to a second ballot, for which a 67% majority was required. If this ballot was not successful, further changes were proposed until a≥50% majority was attained (or the proposal rejected). The points to consider are presented in the wording they were finally voted on (table 1). The results of the respective last ballot are shown as percentage of present members in table 1. Notes captured the contents of the discussions and the reasoning behind each decision to be presented in the comments accompanying the individual items in the manuscript. Data which emerged after the voting process, such as material made public by regulators, were taken into consideration in the manuscript to provide the readers with up-to-date information.
Points to consider for the treatment of patients with immune mediated inflammatory diseases with Janus kinase (JAK) inhibitors
After the face-to-face meeting, the points to consider as agreed by the task force received a final adjudication in terms of level of evidence and strength of recommendation. They were finally subjected to an anonymous vote (by email) on the levels of agreement. Each recommendation received an adjudication on a scale of 0–10, 0 meaning no agreement whatsoever and 10 absolute agreement. The draft of the manuscript was sent to all task force members for their comments which were all considered for the final version prior to submission of the manuscript.
Results
General principles
The task force agreed on four general principles (table 1). The first of these refers to the importance of shared decision making between the patient and the specialist, including information on the benefits and risks of JAKi which is highlighted as principle A. This is in line with various management recommendations but needs to receive special emphasis when a drug or class of medicines is new and long-term experience is still lacking.
The task force further recommends to use these points-to-consider together with general management recommendations for the individual diseases which are usually provided by the respective international or national societies (item B) and also to refer to the product information related to the specific disease to be treated (see below item D).
At outset, the task force decided not to provide ‘recommendations’ for the use of JAKi in treatment algorithms, but rather ‘points to consider’ assisting the clinician when thinking of starting, or having decided to start treatment with a JAKi (principle C). Recommendations may be seen as too directive and would have to be brought into the context of other medications and general treatment strategies and adjusted as new information comes to hand in a rapidly evolving therapeutic area. In contrast, the task force saw its role in elucidating important aspects that should be taken into account when thinking of the prescription of a JAKi. To this end, general principles as well as specific considerations are highlighted as an adjunct to product information (principle D).
Individual points
Six major groups of consideration are highlighted (table 1): indications; dosage and comedications; contraindications; pretreatment screening and risks; adverse events; and laboratory and clinical follow-up. The order within these groups does not relate to any ranking by importance but occurred either by chance or some rationale-based approach to therapies in general.
I. Indications
JAKi have proven efficacious with acceptable safety in patients with a variety of IMIDs. They have received regulatory approval for patients with RA, PsA and UC who have failed prior conventional synthetic DMARD (csDMARD) or bDMARD therapy, and an approval is being sought in further indications, such as dermatological, and interferonopathies. Approval of additional JAKi for IMIDs is expected. At present, individual JAKi have been approved for different diseases and at varying doses, as detailed below.
Treatment dose and comedications in different IMIDs
Treatment doses and comedications may differ between indications (see below) and may have to be adjusted with higher age and organ (hepatic, renal) function impairment. Once the therapeutic target (such as remission) is reached, dose reduction or increase of intervals between doses may be considered; this dose adjustment is not within the label of the JAKi, but similar dose changes outside the label have been suggested for bDMARDs in various recommendations.44 45 In the following, we will address these aspects for the individual IMIDs for which JAKi are approved or may be licensed in the future.
Rheumatoid arthritis
Addition of a JAKi to continued methotrexate (MTX) or other csDMARDs should be considered if the patient tolerates the csDMARD,44 since—just like for all bDMARDs—there is evidence for better efficacy of combination compared with monotherapy, clinically and/or structurally.46 47 Monotherapy of JAKi compared with MTX monotherapy in MTX naïve RA patients failed to show significant structural (though not clinical) superiority for baricitinib47 and—for the primary endpoint—failed to show clinical (though not structural) superiority for filgotinib,48 while combination therapy achieved significant superiority across all outcomes. On the other hand, monotherapy of tofacitinib and upadacitinib in MTX naïve patients had significantly better clinical and structural efficacy than MTX monotherapy,49 50 but neither was investigated in 3-arm trials with an additional combination arm. In contrast, filgotinib was assessed in a 3-arm trial in MTX-naive RA patients; while at both the 100 mg and 200 mg dose filgotinib plus MTX attained the primary endpoint of superiority against MTX monotherapy, filgotinib monotherapy at 200 mg failed to show statistical superiority compared with MTX monotherapy (filgotinib 100mg as monotherapy was not tested), 1 48 None of the JAKi has ever been compared with MTX plus glucocorticoids, the standard therapy recommended by EULAR for over a decade44 which has not been shown to be inferior to bDMARDs plus MTX.51 52 However, a JAKi can be given as monotherapy in case of intolerance or contraindications to MTX and other csDMARDs.
The recommended dose of tofacitinib for RA is 5 mg two times a day in most countries; at this dose tofacitinib was superior to placebo in patients with active disease despite MTX or prior bDMARD therapy, and in a head to head study tofacitinib 5 mg two times a day combined with MTX was non-inferior (but not superior) to adalimumab combined with MTX while monotherapy of tofacitinib 5 mg two times a day failed to show non-inferiority to either combination therapy with tofacitinib plus MTX or adalimumab plus MTX.46 Of note, according to information by regulatory authorities tofacitinib at 10 mg two times a day was associated with an increased venous thromboembolism (VTE) and pulmonary embolism (PE) rate in patients with RA enriched for cardiovascular risk factors,53 54 and a similar warning was also issued for 5 mg two times a day55 (see also below). In addition, the dose should be reduced in patients with a creatinine clearance (CrCl) of <30 mL/min and is contraindicated in patients with severe hepatic impairment (Child Pugh C).
The recommended dose of baricitinib in RA is 4 mg once daily (except for some countries such as USA, Canada and China, where it is 2 mg daily). At the 4 mg dose in combination with MTX, it showed superior efficacy to placebo (or de novo MTX) in all RA patient populations, MTX/csDMARD-insufficient responders (IR), bDMARD-IR or MTX-naïve, respectively; in most countries the approval is for these populations apart from MTX-naïve patients, as combination therapy or monotherapy. A dose of 2 mg once daily is appropriate for patients aged ≥75 years, those with a CrCl of 30–60 mL/min and may be appropriate for patients with a history of chronic or recurrent infections. In a head to head study baricitinib 4 mg per day combined with MTX had superior efficacy compared with adalimumab 40 mg combined with MTX.56 Subanalyses revealed that this superiority was primarily seen for patient reported outcomes, but not for joint counts. In a tapering study patients in long-term low disease activity or remission after 15 months therapy could reduce baricitinib to 2 mg per day; low disease activity (LDA) was maintained at 12 weeks after step down in 83% of patients; 90% of those who flared regained their original response after dose increase.57 Combination of baricitinib with MTX had significantly better structural outcomes compared with MTX alone, but—while monotherapy was similar to combination therapy in terms of clinical and functional outcomes—structural benefit was not significant for baricitinib monotherapy.47
Since the date of the SLR, upadactinib has been approved by FDA and EMA at 15 mg daily. At this dose, it showed superior efficacy to placebo (or de novo MTX) in all RA patient populations, MTX-IR, bDMARD-IR or MTX-naïve, respectively. In most countries, the approval is for these populations except for MTX-naïve, as combination therapy or monotherapy. A upadacitinib monotherapy study in patients with IR to MTX had high response rates but lacked a comparator group of upadacitinib combined with MTX.49 In combination with MTX, upadacitinib 15 mg provided superior efficacy compared with adalimumab plus MTX in a head to head study.58 As in the study of baricitinib versus adalimumab, subanalyses revealed that this superiority was primarily seen for patient-reported outcomes, but not for joint counts.58 No dose adjustment is needed for renal impairment, but with severe hepatic impairment the drug is contraindicated.
More recently, filgotinib was approved at 100 mg and 200 mg doses in Europe59 and in Japan. In contrast, FDA did not approve filgotinib wishing to await data from spermatogenesis safety studies and raising concern about the safety of the 200 mg dose.60 Filgotinib has completed phase 3 clinical trials at 100 mg and 200 mg daily. Data of a study comparing filgotinib plus MTX head to head with adalimumab plus MTX recently became available and revealed non-inferiority for DAS28-C reactive protein (CRP) <3.2 for the 200 mg, but not the 100 mg dose; it was not possible to claim superiority versus adalimumab plus MTX due to the statistical plan.61 Filgotinib has also been studied as monotherapy (see above).
Peficitinib showed significant efficacy on symptoms, signs and structural outcomes of RA in randomised trials including monotherapy and concomitant MTX treatment, in patients with an IR to TNFi and an open label study with etanercept as a safety control. These studies included a majority of Japanese, Korean and Taiwanese patients,62 63 while the difference to placebo was small in a global study where very high placebo response rates were seen.64 Peficitinib is approved in Japan and Korea at 100 and 150 mg daily.
The dose of JAKi should be modified according to patient-specific demographics, comorbidities and/or concomitant medications as per product monograph inserts (see also section on contraindications).
There is no evidence at present that one JAKi is more efficacious clinically, functionally or structurally or safer than another JAKi. While two studies have shown superior efficacy of a JAKi plus MTX compared with an anti-TNF plus MTX,56 58 two other trials have failed to show such effect46 61; moreover, in the trials showing superiority of a JAKi plus MTX to adalimumab plus MTX, the significantly better efficacy was seen for most outcomes, but not for tender and swollen joint counts.56 58 Thus, the relevance of this finding is currently limited; moreover, as of now, no study compared one JAKi with another. On the other hand, several of the above cited studies clearly revealed that JAKi have superior efficacy than TNFi for pain and fatigue, an aspect that deserves further investigation, as it may relate to a hitherto insufficiently recognised and specific mode of action for this drug class.
No studies are yet available in JAKi IR or intolerant patients switching from one JAKi to another JAKi. However, a recent study showed efficacy of anti-TNF therapy after IR to a JAKi,65 and safety regarding switch from a bDMARD to a JAKi without washout, information that was missing hitherto.
Regarding dose reduction in patients with RA in sustained Clinical Disease Activity Index (CDAI) or Boolean remission on background csDMARD, trial evidence is currently confined to dose reduction for baricitinib.57
Psoriatic arthritis
Currently, only tofacitinib is approved for PsA; the licensed dose is 5 mg two times a day. The clinical trials demonstrated efficacy in patients with prior IR to csDMARDs66 and TNFi.67 The efficacy in PsO is described below.
Since the closing date of the SLR, two phase 3 trials of upadacitinib were completed successfully, showing efficacy regarding main outcomes of PsA, also when compared with adalimumab (non-inferiority for the ACR20 response with the 15 mg and superiority with the 30 mg dose).68 69
Filgotinib at 200 mg daily showed efficacy in a phase 2 trial70 and phase 3 data are awaited.
Ankylosing spondylitis
Tofacitinib demonstrated significant efficacy at 12 weeks for signs and symptoms in patients with highly active AS (by modified New York criteria) refractory to NSAIDs in a phase 2 dose-ranging placebo-controlled RCT. The highest Assessment of SpondyloArthritis international Society (ASAS) 20 response was observed at 5 mg two times a day, especially in patients with both elevated CRP and evidence of MRI inflammation in the sacroiliac joints.71 A dose-dependent effect for clinical response was not evident. Separation from placebo was observed at 4–8 weeks suggesting a slower onset of response than seen with TNFi.
Filgotinib at 200 mg once daily has been assessed in patients with active AS refractory to NSAIDs and TNFi (10%) in a 12 week phase 2 placebo-controlled RCT.28 Significant benefit for disease activity, assessed by the AS Disease Activity Score (ASDAS), was evident by week 1 and major improvement in ASDAS was noted in 33% versus 2% of patients on filgotinib and placebo, respectively, at 12 weeks. For most outcomes separation from placebo was observed at 4–8 weeks.
Upadacitinib at 15 mg daily was assessed in a 12-week phase 2/3 placebo-controlled trial that recruited patients with active AS refractory to NSAIDs.72 Significantly more patients had an ASAS 40 response at week 14 in the upadacitinib versus the placebo group (52% vs 26%) and this was observed at the first postbaseline visit at week 2. Other outcomes including MRI spine and sacroiliac joint inflammation, were also superior for upadacitinib, just like for tofacitinib and filgotinib.
Overall, the 12-week phase 2 data support the efficacy of JAKi for a variety of disease outcomes relevant to AS to a degree comparable to TNFi while the pattern of AEs and changes in laboratory outcomes were similar to those reported in previous studies in other indications.
Dermatological diseases including PsO
Nine different JAKi and three selective TYK2 inhibitor have been evaluated in PsO; none of them has been approved for this indication to date except for occasional individual countries, such as Russia (tofacitinib).
Tofacitinib was tested in one phase II, four phase III and one long-term extension study, the results of which were recently summarised.73 Tofacitinib 5 and 10 mg two times a day showed superiority over placebo for all efficacy endpoints at week 16, with response maintained for 52 weeks of continued treatment. The Psoriasis Area and Severity Score (PASI) response, however, appeared numerically lower than that typically seen for bDMARDs such as IL-12/23 or IL-17 inhibitors. Tofacitinib improved patients’ quality of life and was well tolerated. With the exception of herpes zoster, rates of safety events of interest were similar to those in the published literature and healthcare databases for other systemic PsO therapies. Tofacitinib 10 mg two times a day demonstrated greater efficacy (PASI75 at 12 weeks: 43% in MTX-IR and 21% in TNFi patients) than 5 mg two times a day. An additional phase IIa study evaluating topical application of tofacitinib in mild-to-moderate PsO found significant clinical improvement over placebo treatment after 4 weeks.74
Baricitinib was studied in a phase IIa dose-ranging study, using once daily dosing over 12 weeks. A statistically significant difference among patients exhibiting at least a 75% improvement in their PASI (PASI75) when compared with placebo was observed for patients receiving 8 or 10 mg daily (43% and 54% vs 17%),75 a much higher dose than approved for RA and, again even at this dose the skin response is numerically lower than reported for several of the more recently approved bDMARDs; for example, for IL-12/23 inhibitors the PASI75 amounted to about 70% and 80% for ustekinumab and guselkumab, respectively,.76 77 and to 89% on IL-17 inhibition.78
For the JAK1 inhibitors abrocitinib, itacitinib and GSK2586184 as well as the JAK1/3 inhibitor peficitinib, data in the public domain are equally available from one phase II study each. 60% of patients receiving the most effective dose regimen (200 mg twice daily) of abrocitinib experienced a 75% improvement of the PASI.79 Itacitinib showed significant improvement in the Physician Global Assessment (PGA) after 4 weeks of treatment with 600 mg once daily versus placebo,80 while GSK2586184 at 400 mg once daily yielded a 75% PASI improvement in 57% of patients after 12 weeks,81 and patients treated with 50 mg of peficitinib once daily benefitted from improved PASI, PGA and reduced body surface area affected.82
Another JAKi, ruxolitinib, was tested in two topical formulations containing 1% and 1.5% of ruxolitinib, respectively, versus placebo and two active comparators, namely calcipotriene 0.005% cream and betamethasone doproprionate 0.05% cream. A statistically significant difference versus the vehicle was observed after 4 weeks of treatment for the ruxolitinib 1% group, with comparable efficacy of ruxolitinib formulations with the active comparators.83
For two additional JAKi, no peer-reviewed publicly available data have been published so far.
Finally, a phase II study evaluating the TYK2 inhibitor BMS-986165 was recently published.41 Doses ranging from 3 mg every other day up to 12 mg daily were studied and compared with placebo. BMS-986165 at doses of 3 mg daily and higher was found to result in greater clearing of PsO than did placebo over a period of 12 weeks, with PASI75 responses up to 75% in the highest dose group. Data for two additional TYK2 inhibitors await peer-reviewed publication.
Evidence for therapeutic efficacy of JAK-inhibitors or TYK2-inhibitors has also been suggested in several other immune-mediated inflammatory dermatoses, including atopic dermatitis, alopecia areata, vitiligo,84 palmoplantar pustulosis and a case of a mucocutaneous disease called idiopathic erythema multiforme associated with a mutation in TRPS1 and JAK-STAT activation.85
Taken together, PsO belongs to a group of chronic inflammatory skin diseases for which inhibition of JAKs or TYK2 has shown clinical efficacy. However, the extent of efficacy observed and the safety profile of the JAKi has so far not led to drug authorisation by EMA or FDA, but there is considerable interest around TYK2 inhibition, given the absence of some safety issues linked to non-selective JAKi, as well as regarding indications other than PsO, namely AD, where there are still far less options available for systemic therapies.
Inflammatory bowel disease
Tofacitinib is indicated for the treatment of adult patients with moderately to severely active UC who have had an inadequate response, lost response or were intolerant to either conventional therapy or a biologic agent.34 Tofacitinib 5, 10 and 15 mg two times a day showed significant efficacy for remission (Mayo score) in patients who were cs- and bDMARD-IR.86 The recommended dose is 10 mg given orally twice daily for induction for 8 weeks (or 16 weeks if adequate benefit is not achieved) and 5 mg given twice daily for maintenance (in TNFi-IR patients 10 mg two times a day maintenance may be used). The 10 mg two times a day dose was associated with VTEs and PEs in patients with RA53 (see also below).
Upadacitinib and peficitinib similarly have shown efficacy in phase 2 trials in csDMARD and bDMARD-IR patients in dose-ranging studies of UC.87 88
In CD, tofacitinib at 5–15 mg two times a day has shown no significant efficacy in induction and maintenance of Crohn's disease activity index remission (<150) compared with placebo.35 However, selective JAK1 inhibition by filgotinib showed increased remission rates in patients with moderate to severe CD.36 Moreover, upadacitinib also showed promising results in a phase II trial in CD37 and larger phase III trials have been initiated. Collectively, these findings hold promise for these agents as clinical therapeutics in IBD which await corroboration in ongoing phase III trials.
Other diseases
Baricitinib was investigated in a phase 2 trial of systemic lupus erythematosus and demonstrated significant efficacy at 4 mg but not 2 mg compared with placebo.89
Other indications for which JAKi are being evaluated include non-infectious uveitis, CANDLE syndrome and other interferonopathies, including USP18 deficiency.90–92 The reader is referred to the SLR manuscript.43
II. Treatment dose and comedication
1. Use the dose recommended for the specific disease.
The dosing of individual JAKi in the various diseases has been addressed in the previous section (see above).
2. Dose adjustments due to drug interactions
Tofacitinib is metabolised by the hepatic cytochrome P (CYP) 450 pathway which leads to drug interactions with inhibitors such as ketoconazole and promoters such as rifampicin, necessitating dosage adjustments, although it is also 30% renally excreted. In contrast, baricitinib is 70% renally excreted. Filgotinib is metabolised by hepatic carboxylesterases and has a major metabolite GS-829845 which is a pharmacologically active, selective inhibitor of JAK1, but is 10–20 times less potent than the parent compound. Upadacitinib predominantly undergoes hepatic oxidation with minor CYP metabolism and peficitinib undergoes hepatic conjugation. Organic anion transporter 3 inhibitors, such as probenecid, interact with baricitinib requiring a dose reduction to 2 mg per day (with normal renal function). Rifampicin when used in latent tuberculosis (TB) prophylaxis or therapy for active TB increases hepatic metabolism of tofacitinib and upadacitinib so that a dose increase of the latter has to be considered. Ketoconazole has the opposite effect, inhibiting tofacitinib and upadacitinib metabolism so a dose reduction is suggested. Dose adjustments due to hepatic or renal impairment are discussed below.
The dosing of the individual agents and their metabolisation are summarised in table 2.
3. Comedication
Comedication has been addressed in the previous section, including addition of JAKi to pre-existing csDMARDs as combination therapy.
4. Consider dose reduction in sustained remission on background therapy
This aspect has also been addressed in the previous section. For dose adjustments due to impaired organ function see below.
Dosing and metabolisation of the different Jakinibs
III. Contraindications that should be considered
Contraindications are primarily related to the adverse event and the pharmacokinetic/pharmacodynamic profile of the various JAKi.
Severe active infections, acute or chronic, including latent TB and opportunistic: these infections can be seen in patients treated with JAKi.93 Serious infection rates in RA and PsA studies of tofacitinib, baricitinib and upadacitinib were comparable to adalimumab with higher rates occurring at higher doses.12 93 Tofacitinib treated RA patients above 65 years (with cardiovascular risk factors) exhibited a higher rate of serious infections compared with TNFi treated patients and according to EMA tofacitinib should be used in these patients only if there is no other alternative.94 A recently published post hoc analysis of RA trial data that included an adalimumab comparator found similarly increased risks for serious infections among the elderly, particularly among those using 10 mg two times a day tofacitinib. Risk elevations as compared with younger patients were similar for those using adalimumab and tofacitinib 5 mg two times a day, but several fold higher for those using 10 mg two times a day.95
Malignancy: using a JAKi in this situation should be a shared decision with the patient given timing of past malignancy, uncontrolled malignancy or ongoing treatment with chemotherapy including checkpoint inhibitors. Thus far, patient registries and clinical trial data have demonstrated no malignancy signal. There are no data to suggest that prior malignancy is problematic with JAKi therapy, but most studies excluded patients with malignant disease up to 5 years prior to enrolment.
Severe organ dysfunction: With severe hepatic disease (Child-Pugh C), JAKi should not be used. With respect to severe renal disease (CrCl) <30 mL/min), a reduction in dosage is recommended for tofacitinib to 5 mg once daily; baricitinib is not recommended if CrCl is <30mL/min. With CrCl 30–60 mL/min baricitinib should be used at 2 mg daily. No dosage reduction is currently recommended for other JAKi.
Pregnancy and lactation: Limited data are available and contraception while taking JAKi is advised for both female and male patients in the absence of adequate data. Tofacitinib has been shown to be teratogenic in rats and rabbits, and to affect parturition and peri/postnatal development.96
Filgotinib reduces spermatogenesis in a dose-dependent manner in animal studies; to date, this has not been observed in humans, but a definitive study evaluating this question is currently underway97 so this can then be taken into account appropriately in male patients.
These agents have short plasma half-lives but a gap of 4 weeks is recommended after the last dose if future pregnancy is being contemplated. It is not known whether tofacitinib is secreted in human milk, but it is secreted in the milk of lactating rats. A risk to the breast-fed child therefore cannot be excluded. As a precautionary measure, the use of tofacitinib during breast-feeding is contraindicated.
A study of a small number of patients with UC taking tofacitinib observed healthy newborns, no foetal deaths or congenital malformations and spontaneous abortions appeared consistent with background risks in the USA.98 Similar data exist for RA and PsO.99
History of VTE events: in patients with a history of thromboembolic events initiation of a JAKi should be carefully evaluated based on the increased rates of VTEs in patients at risk for these events (see below under risks and adverse events). Increased VTEs, especially PE, have been observed in patients with cardiovascular risk factors treated with 10 mg tofacitinib two times a day53 54 indicating that also these (arterial) risks require consideration. Patients with recurrent thromboembolic events will usually receive anticoagulation treatment likely counteracting the risk.
The safety and efficacy of JAKi is under investigation in juvenile patients but has not yet been established in persons <18 years of age. For restrictions regarding patients >65 years of age see below.
Finally, JAKi have not been studied and, therefore, are not recommended in combination with bDMARDs or potent immunosuppressive agents such as cyclosporine or tacrolimus because of the possibility of increased immunosuppression and increased risk of infection or lymphoma.
IV. Pretreatment screening and risk assessments
History and physical examination: Important patient details to obtain before starting therapy include a history and risk estimation of VTE, infections, TB, risk factors for hepatitis B and C, as well as usual medical considerations such as comorbidities, cardiovascular risk factors and concomitant medications of relevance, for example, Cox 2 inhibitors, prednisone doses >7.5 mg daily or oral contraceptives.
The recommendation for patients over 70 years of age is a baricitinib dose reduction to 2 mg daily due to age-related reductions in renal function. Moreover, EMA has restricted the use of tofacitinib in people older than 65 years also due to an increased risk of serious infections.94 No dose reduction is recommended for modest renal impairment with upadacitinib and filgotinib therapy.
Baseline skin check for non-melanoma skin cancer (NMSC) in patients at risk and chest X-ray is also recommended, unless recently performed.
Routine laboratory testing that includes a full blood count (including a differential white cell count), liver enzymes (in particular transaminases), and renal function tests are recommended before starting JAKi. Baseline lipid levels are suggested unless recently checked. No creatine phosphokinase (CPK) testing is needed.
Hepatitis B virus (HBV) testing for anti-HBs, anti-HBc, and HBsAg is recommended in all patients. Patients with evidence of chronic HBV infection (ie, positive HBsAg) should avoid JAKi or treatment with biologics if possible. If not possible, then concomitant treatment or prophylaxis with an anti-viral (eg, entecavir, tenofovir or tenofovir alafenamide) should be undertaken alongside consultation with a hepatologist.100 For patients with evidence of prior HBV exposure (positive HBc antibody) and no evidence of active viral replication (ie, negative HBsAg), a baseline HBV DNA should be obtained to rule out occult active HBV infection. If positive, then patients have active HBV and should be managed according to the above. The main virological event of concern in these anti-HBc positive patients is HBsAg reappearance (seroreversion), consistently associated with hepatitis flare; HBV DNA detection (without HbsAg) leads to seroreversion and hepatitis in 50% of cases.100 If HBV-DNA negative, then such patients can start JAKi and should be routinely monitored for HBV DNA and HBsAg reappearance (seroreversion) in line with respective national recommendations for TNFi. If HBV DNA or HBsAg subsequently turns positive during monitoring, then the patient should be managed as above with referral to a hepatologist for treatment. The JAKi should be temporarily stopped until full evaluation can be made. Concurrent treatment with an antiviral is possible, and the JAKi can be reinstituted once anti-viral therapy has been started.101
Hepatitis C virus (HCV) antibody testing is recommended and should be further assessed if positive, that is, HCV RNA testing. If positive, then the patient has active HCV and should be referred for treatment. In such case, JAKi should be withheld until HCV treatment has been completed.
HIV testing is recommended for those with HIV risk factors.
As the risk for TB-reactivation with JAKi is similar to that for TNFi, screening for TB is recommended, unless already done prior to bDMARD commencement without a risk of exposure since then. All patients in JAKi phase 3 studies were screened for TB and patients with active TB excluded while patients with latent TB were commenced on anti-TB therapy and included. Cases of TB were noted with JAKi more commonly than with placebo in pivotal trials, with at least some cases occurring in endemic areas likely representing newly acquired infection rather than reactivation of prior infection.12 93
Vaccination status should be sought. Country and regional vaccination guidelines should be followed. EULAR has recently updated its vaccination recommendations for patients with autoimmune diseases.102 In addition to Herpes zoster reactivation, Herpes simplex and cytomegalovirus reactivation may also occur. HPV reactivation is not known to occur, but has not been evaluated systematically.
Herpes zoster reactivation: A history of varicella or zoster infection or immunisation should be obtained. Herpes zoster reactivation is clearly increased under JAKi with incidence rates (IRs) between 3–4 (Western Europe, USA, Australia) and 9 (Japan, Korea) per 100 patient-years compared with 2–3 per 100 patient-years for TNFi. Risk factors include age, female gender, prednisolone >7.5 mg per day, infection and hospitalisation.11 12 103–106 As for serious infections, there is also a dose response for Herpes zoster reactivation. The reactivation is likely based on the mode of action of JAKi blocking interferon pathways. If a patient develops Herpes zoster, JAKi treatment should be temporarily interrupted until the episode resolves. A small proportion of patients can develop recurrent zoster. Antiviral prophylaxis could be considered in such individuals.
Evidence for the efficacy of the live Zostavax vaccine is questionable and as a live attenuated vaccine it necessitates a delay of 3–4 weeks postvaccination before starting a JAKi; further, a single missed dose of MTX could be considered, since MTX may blunt the antibody response,107 but this approach is not evidence based. In a live zoster vaccination study, zoster IRs at follow-up were numerically similar in the tofacitinib 5 mg and adalimumab and MTX arms but higher rates were seen for the combination of tofacitinib 5 mg two times a day with MTX.108 Notably, zoster rates at follow-up were generally similar in vaccinated versus non-vaccinated patients, but further studies are clearly needed. While the vaccination resulted in reasonable immune responses, 1 patient developed zoster infection having had no prior immunity.109
A new zoster vaccine has been more recently approved; being a nonlive vaccine, it is not contraindicated in patients receiving immunosuppressive or immunomodulating agents, but currently there are no data on safety and protective immunogenicity of this vaccine in patients treated with JAKi. As studies are underway, these questions should be resolved soon. The safety of the inactivated zoster vaccine (Shingrix) has been suggested by a small open label study of 400 patients with RA (no zoster activation, 6.7% disease flares, mostly mild, self-limiting, and not requiring therapeutic change), but efficacy and immunogenicity of the vaccine in this setting is unknown.110
Risk factors for VTEs111 ,112 should be considered by history and a potential clotting abnormality should be pondered in patients with a history of VTEs in whom such assessments have not yet been done. While these events are rare, the risks are increased in patients with prior VTEs; with increasing age (patients older than 65 years are at higher risk for having VTEs with tofacitinib); obesity (people with obesity have two times the risk of VTEs as people with normal weight, and the higher the weight, the higher the risk); prolonged immobility (ie, long travel, lower-extremity paralysis due to spinal cord injury, trauma with reduced mobility); hereditary (ie, factor V Leiden, prothrombin mutation 20210, etc) and acquired (ie, antiphospholipid syndrome, malignancy) thrombophilia; Cox 2 inhibitor therapy113 114; prednisolone of ≥7.5 mg/d and above; major surgical interventions, such as neurosurgic, urologic, gynaecologic and orthopedic surgery. Interestingly, a recent study from Sweden suggested that VTEs are significantly related to disease activity with an adjusted RR of 1.99 during high, 1.45 during moderate and 1.11 with low RA activity compared with remission.114 ,115 This potential relation between VTE rates and RA disease activity remains to be fully elucidated. For more details regarding VTEs and PEs see below under adverse events.
V. Adverse events
Adverse events are mainly related to the inhibition of cellular pathways and include those already mentioned above under risks. However, several other adverse events need more detailed consideration.
Serious infections including opportunistic infections such as TB and others, as well as reactivation of Herpes zoster and other viruses can occur. The IR of Herpes zoster reactivation amounts to about 3–4 compared with placebo (IR=1).115 116 Their frequency is dose and co-medication dependent and the reactivation of Herpes zoster is more frequent than on bDMARDs and especially frequent in Japan and Korea. Moreover, EMA (but not FDA) has restricted the use of tofacitinib in people older than 65 years due to an increased risk of serious infections.94 Herpes zoster was also seen with baricitinib and less commonly with upadacitinib. It is also listed as an important potential risk for filgotinib by EMA, and while they state that no signal for varicella zoster infection has been detected in the filgotinib RA clinical trial program, the agency requests further evaluation by additional pharmacovigilance activities.117
Malignancy: The overall rates are not increased except for the risk of NMSC which might be elevated and, therefore, the task force recommends regular skin examinations, especially in countries with increased risk of NMSC, such as Australia. The task force also felt that current malignancy (except NMSC and cervical carcinoma in situ undergoing treatment) may be a contraindication for JAKi, but as stated previouly, this should be a shared decision making with the patient.
Anaemia and cytopenias: Anaemia of chronic disease, as usually seen in most IMIDs, does not improve on the group level with all JAKi except for filgotinib, and in some patients the pre-existing anaemia may deteriorate, presumably due to JAK 2 inhibition; JAK 2 is involved in EPO signalling (see figure 1). Cytopenias may occur but were not more frequent than on placebo,12 although with all JAKi a few patients may exhibit neutropenia and/or lymphopenia.
VTE/pulmonary embolism (PE). Across indications, in randomised controlled trials and long term extensions of tofacitinib followed for up to 9.5 years, no increased risk of VTE for the 5 mg bd dose has been seen.11 54 However, in a still ongoing safety study of patients with RA enriched for cardiovascular risk factors, a statistically significant PE imbalance for tofacitinib 10 mg bd as compared with 5 mg two times a day was demonstrated with an absolute IR of 0.5 for 10 mg and 0.3 for 5 mg53–55; compared with TNFi (absolute IR of 0.1) which were investigated as a control arm, the PE risk thus being about threefold higher for the 5 mg dose and about sixfold higher for the 10 mg dose.54 94 In this same study, as reported by the EMA, VTE without PE were somewhat numerically higher with tofacitinib than TNFi but the difference was not statistically significant. Since this is an ongoing study, the full data for the 5 mg dose will have to be awaited for a full assessment of the VTE risk, but the 10 mg two times a day dose was discontinued in this study. A recent analysis of VTE/PE in clinical trials of UC in which most of the patients had been treated with a dose of 10 mg two times a day reported that during the placebo controlled period no UC patient had a VTE or PE and 1 VTE and 1 PE each were seen in placebo treated patients.118 During the long term, open-label extension comprising about 2400 patient-years of exposure, 1 patient had a VTE and 4 had PEs, all with risk factors for these events.118 The recent EMA assessment provided evidence for an increased PE risk at the 5 mg and especially the 10 mg two times a day dose of tofacitinib.55 The FDA has not made a final determination and is awaiting the final, adjudicated results of the study.
Baricitinib at 4 mg had an imbalance in VTEs compared with the control arms (placebo or adalimumab) in the controlled period of RA trials; this has not been observed with the 2 mg dose.49 119 Risk factors were age, high BMI, immobilisation, surgery, use of Cox-2 inhibitors and a history of prior VTEs; the risk may be up to 10-fold for patients with a history of VTEs and twofold for those patients taking Cox2 inhibitors.120 Subsequently, there was no increase in risk when patients were transitioned from placebo or MTX to baricitinib as well as across long-term extension studies over 6 years but VTEs in the LTE were observed equally with 2 and 4 mg.
Numerically increased rates of VTEs have also been observed in the double blind phases of upadacitinib trials, primarily with the 15 mg once a day dose, although not in the head-to-head trial against adalimumab.49 58 119 With respect to filgotinib, EMA regards VTE as a potential risk, but the agency also concluded that no increase in reports of VTEs was seen for filgotinib (100 mg and 200 mg doses) compared to placebo or comparators (MTX, ADA). Importantly, however, additional data by pharmacovigilance activities have been required by EMA.
Taken together, these observations elicited warnings (in some countries "black box" warnings) for VTE in the labels of all approved JAKi, plus additional warnings issued by the regulators (see above). In particular, the EMA recommends the use of tofacitinib in patients with RA ‘above the age of 65 only when there is no alternative treatment’.53 55 Such age considerations have not been set in place for other JAKi that have similar VTE warnings in their label; however, data on outcomes studies in patients with cardiovascular risk factors are not available for those other agents. VTEs on baricitinib also occurred in patients with risk factors, such as obesity, and several continued therapy, although mostly under anticoagulation.120
While the overall risk of VTE is age dependent and in the order of 1:100–1:1000 (occurrence rate 0.25/100 patient years), this risk is about doubled in patients with RA.55 Further research is needed to delineate the mechanisms how JAKi increases VTE rates (and how this compares to patients with RA in general), while we also lack understanding how glucocorticoids, Cox2 inhibitors, oral contraceptives, tamoxifen, thalidomide, antipsychotics elevate VTE risk. In any event, careful consideration should be given as whether or not to start a JAKi in any patient who may be at risk for a VTE.
With respect to major adverse cardiovascular events (MACE) across indications, in randomised controlled trials and long-term extensions no increased risks have been observed. As indicated above, a long-term study of tofacitinib in RA patients with cardiovascular risks is ongoing and has hitherto not shown evidence for an increase in MACE (regarding increase in VTEs see above).
Laboratory abnormalities without clinical sequelae in the majority of patients: CPK elevations are occasionally seen without weakness, thus far with occasional myalgia,121 122 but usually without clinical repercussions, although one patient has had rhabdomyolysis.123 Thus, in the rare event of symptoms, CPK should be tested, although in general this is not necessary. While the underlying cause is unknown and there have been suggestions this may be due to a renal tubular effect, there are some data suggesting this effect might be due to restoration of muscle development with associated CPK elevations (an event that is suppressed by oncostatin M whose signalling depends on JAKs).124 125 Creatinine increases have also been observed but without organ dysfunction or other clinical sequelae, such as hypertension.
Finally, gastrointestinal perforation has been reported in clinical trials and may be a risk of bariticitinb and tofacitinib126 (and possibly other JAKi). Thus, JAKi should be used with caution in patients who may be at increased risk for gastrointestinal perforation (eg, patients with a history of diverticulitis and taking concomitant NSAIDs or glucocorticoids). Patients presenting with new onset abdominal signs and symptoms should be evaluated promptly for early identification of gastrointestinal perforation knowing fever and elevation of acute phase reactants may be blunted by JAKi therapy.
VI. Laboratory and clinical monitoring during follow-up
As a minimal laboratory monitoring during follow-up, the task force recommends measurement of full blood count and differential, transaminases, renal function, at 1 month and 3 months and then periodically such as every 3 months plus lipid levels just at 3 months.
Blood count: Haemoglobin change of less than or equal to 20g/L decrease and haemoglobin levels greater than or equal to 90g/L do not require dose adjustment. Greater than a 20g/L decrease or a haemoglobin of less than 80g/L (confirmed by repeat testing) should lead to dose interruption until haemoglobin values have normalised. Filgotinib leads to small dose dependent average increase in haemoglobin levels, compared with all other JAKi.
Absolute neutrophil counts over 1000/mm3 require no dose adjustment, however, a count of 500–1000/mm3 on two sequential measures suggest dose reduction or temporary cessation until count above 1000/mm3 when JAKi can be recommenced.
Absolute lymphocyte counts over 750/mm3 require no dose adjustment, a count of 500–750/mm3 on two sequential measures suggests a dose reduction or temporary cessation until the count is greater than 750/mm3 to allow recommencement. There is some evidence that lymphocyte counts below 500/mm3 significantly increase the risk of opportunistic infection.
Liver function tests: Transaminases should be periodically monitored. Tofacitinib should not be used in severe hepatic impairment (Child Pugh C) nor should upadacitinib. Mild hepatic impairment (Child Pugh A) requires no dose adjustment. In case of moderate hepatic impairment (Child Pugh B) the tofacitinib dose should be reduced to 5 mg once a day.
Renal function: Creatinine should be assessed periodically. In mild to moderate chronic renal impairment (CrCL 50–80 mL/min) no dose adjustment is needed; with CrCL 30–60 mL/min, baricitinib should be reduced to 2 mg daily. With severe renal impairment (CrCl <30 mL/min) tofacitinib dose should be reduced to 5 mg once daily and baricitinib not used at all.
Acute phase reactants: For evaluation and definition of response be aware that CRP and ESR may be reduced independently of reduction of disease activity and, therefore, consideration should be given to the use of disease activity scores that do not include inflammatory markers (such as CDAI in RA; see below under 3).
Lipid levels should be assessed approximately 3 months after JAKi commencement and if increased should be managed according to national guidelines.
Consideration should be given to an annual formal skin check as evidence suggests an increased risk of NMSC with tofacitinib, possibly due to prior exposure to MTX and TNFi.127
Disease activity should be monitored regularly using validated composite measures of disease activity that include joint counts in order to assess if improvement by >50% was seen within 3 months and the treatment target by 6 months (treat-to-target),128 129 in line with current management recommendations for RA and PsA,2 44 and in line with recommendations for other IMIDs, respectively. It should be borne in mind that acute phase reactant levels may be reduced by JAKi independent of clinical improvement and, therefore, scores that are heavily weighted on acute phase reactants, such as the DAS28, should not be used for follow-up.130
Consideration of patient preferences
In rheumatology, there is still a substantial number of patients with suboptimal outcomes or who are faced with uncontrollable disease symptoms. They fail to respond adequately to existing DMARDs. Therefore, the advent of DMARDs with a new mode of action is welcome. The oral route may enable some patients to become more independent from hospital or health professionals compared with subcutaneous injections or infusions and also appeals to those with a needle phobia; on the other hand, some biological agents are only administered monthly or even less frequently and this may be seen as an advantage compared with taking a drug once or twice daily. Cost considerations are an overarching principle in RA treatment recommendations and thus part of treatment decisions; while the costs of JAKi are currently usually higher than those for biosimilars, this may change once these drugs become generic. Careful consideration of initiation and open communication with the patient are warranted. The prescription of JAKi may not be at the expense of attention to safety risks and must be in line with existing specialty guidelines for management and good clinical practice which also includes the need for regular laboratory monitoring even in patients receiving JAKi monotherapy. These points to consider contain important information for patients. A patient version or a decision tool will support patients to weigh potential benefits, harms and their personal goals and preferences, and subsequently strengthen their role in the decision-making process.
Research agenda
The committee felt that many questions remained open and needed to be addressed in future research in both adult and paediatric populations. These questions are pertinent to all JAKi and are presented in box 1.
Research agenda
What is the efficacy and safety of switching between JAK-inhibitors in non-responders or due to lack of tolerability?
What are the predictors of response to JAK-inhibitors as compared to other DMARDs used for RA?
What is the effect of JAK-inhibitors on comorbidities of IMIDs including cardiovascular disease and osteoporosis?
Is VTE a class effect or a JAK inhibition effect and what is the mechanism of VTE? What is the actual risk of VTE when treating with a JAK-inhibitor? Is the effect confined to RA or observed in other indications?
What is the long-term safety from real-world data for JAK-inhibitors? For which patients should JAK inhibitors be contraindicated on basis of risk (particularly for VTE), and should prophylaxis be considered?
What is the safety of JAK-inhibition in patients with prior, current or who develop a malignancy whilst on therapy?
Are JAK-inhibitors effective and safe as therapy for autoimmune diseases induced by checkpoint inhibitors in patients with malignancy?
How safe are JAK-inhibitors in Hepatitis B, C, SARS-CoV-2 infected patients and also other viral infections?
How safe are JAK-inhibitors in pregnancy and lactation? What should be recommended if a woman taking a JAK-inhibitor becomes pregnant?
Safety of JAK-inhibitors in elective surgery—should they be discontinued and if so for how long and when should they be restarted?
What is the efficacy of JAK-inhibitors in extra-articular RA manifestations including vasculitis, nodulosis, overlap syndromes?
What is the efficacy of JAK-inhibitors in connective tissue diseases such as SLE, inflammatory myositis and systemic sclerosis?
What is the efficacy and safety of combination therapies with JAK-inhibitors and bDMARDs in patients with severe RA or other diseases?
What are the molecular in vivo down-stream effects of JAK-inhibition in the setting of individual diseases?
What are the differences between different JAK-inhibitors regarding efficacy and safety?
What is safety of JAK inhibitors in patients over 65 years?
Discussion
Similar to the situation with bDMARDs 15–20 years ago, real-world experience with JAKi is limited. Therefore, this task force was formed which consisted of experienced clinical trialists and people involved in treating patients with IMIDs across several medical areas and across nations and continents as well as patients and a health professional. The task force set out to provide the readers with comprehensive guidance on the use of this novel class of targeted therapies regarding efficacy and safety, based on evidence and complemented by expert opinion. In this consensus statement points to consider are provided for the use of JAKi across IMIDs for which they are approved or may be approved in the near future.
The consensus statement is designed to support physicians and other health professionals treating patients with IMIDs as well as patients themselves and other stakeholders, such as hospital administrators and payers, with an up-to-date summary on the thoughtful application of JAKi. Where there is occasional redundancy in the paper, it derives from the fact that certain pieces of information relate to more than one chapter of this consensus statement, thus allowing readers who only focus on selected portions to obtain pertinent information.
Currently baricitinib, filgotinib (in Europe and Japan), peficitinib (in Japan), tofacitinib and upadacitinib are licensed for one or more autoimmune inflammatory diseases. The consensus statement is primarily based on the evidence derived by an SLR43 from clinical trials and some observational studies, whereby safety aspects can currently be primarily or solely derived from information of the controlled and extended trial periods of the drugs.
Indeed, efficacy data from comprehensive clinical trial programmes but hardly any long-term registry data from clinical practice are available on safety aspects . However, trial efficacy and safety data are constantly being expanded, as are the indications. At present five available JAKi are approved for use in RA patients, but tofacitinib is already licensed for PsA and UC and other compounds will also likely receive approval for a range of indications. Thus, JAKi may arrive at a similarly broad or even broader list of indications across IMIDs as TNF-blockers. However, their broad efficacy is unrelated to inhibition of TNF signalling, but rather due to the fact that the intracellular blockade of JAKs relates to cytokines that are distinctly involved in different IMIDs, such as IL-6 in RA, IL-23 in PsA, PsO and IBD, or interferons in other diseases. Moreover, even if none of the cytokines activating the JAK-STAT pathway is known to be of significance in the pathogenesis of a particular disease, such as axial spondyloarthritis, it is possible that there are synergistic inhibitory effects by interfering with signalling of several cytokines that individually are only minimally pathogenetically relevant, culminating in clinical efficacy. Moreover, JAKi also interfere with the consequences of JAK activation induced by cytokines that do not directly use the JAK-STAT pathway for signalling, such as TNF, which can activate IL-6 and interferons downstream of their primary effects, thus affecting various pathogenic pathways.39
There are some differences between the drugs which are due to different selectivities regarding JAKi when looking at both in vivo and in vitro data, spanning from predominant JAK1 inhibition (filgotinib) to pan-JAKi (peficitinib, tofacitinib); these may translate into differences in reversible cytokine inhibition over the dosing period. These differences will be dose dependent19 and may be reflected in variability in safety but also aspects of efficacy.
Recommendations on indications and dosages can easily be derived from the clinical trials and labels of the respective drugs as stipulated by regulators, but the presumably more important items within this consensus statement relate to contraindications, pretreatment screening, safety and risks as well as monitoring and follow-up examinations. All these items have been addressed. The recommendations may change once further pharmacovigilance and registry data become available. They may also change once more information becomes known regarding pathways to disease or pathways leading to adverse events. Of note, the task force was informed by, and developed its recommendations based on, a detailed SLR evaluating studies that were published until the end of 2018; however, since then additional safety aspects became known from information provided by regulators, but the trial(s) on which this new safety information is based have not yet been published. Thus, the task force went beyond the data provided in the SLR and addressed publications and regulatory communications that appeared after the end of the SLR period to provide readers with the most up-to-date material.
Among the adverse events, some have been expected from knowledge regarding blockade of cytokines that use JAK-STATs for signalling, such as an increased risk of serious (including opportunistic) infections. Others go beyond expectations but are explained by the pharmacologic effects of the drugs, such as the increase in herpes zoster rates. The failure to reverse anaemia of chronic disease would also be in line with expectations based on inhibition of JAK2, and this conclusion is confirmed by the improvement of anaemia when a more selective JAK1 inhibitor is applied.131 However, even if relatively rare and associated with known risk factors, the occurrence of VTEs and PEs is an unexpected and hitherto unexplained event requiring further information and elucidation. It is not clear if idiosyncratic platelet activation, changes in procoagulant or fibrinolytic activity, or abnormal endothelial activation might be involved. However, other tyrosine kinase inhibitors may activate procoagulant activity132 which might be related to changes in lipids or lipoprotein levels.133 During the deliberations of the task force, comments arose that the control arms of some pivotal studies had an unusually low IR of VTEs; nonetheless, it is the nature of randomised controlled trials that the risk should be balanced across study arms and if one arm differs, one may conclude that the results are a consequence of the respective treatment. Further research activities in this area are urgently needed.
Since the time of the SLR, the task force meetings and the start of preparing this manuscript, the COVID-19 pandemic has struck the world. Many patients with IMIDs who are treated with bDMARDs or JAKi have contracted this viral disease. Currently, there is insufficient knowledge about the risks (or potential benefits) of immunomodulating drugs in these patients in either altering susceptibility to infection, or in determining disease progression once infected with SRAS-CoV2. Case reports and individual centre’s experiences do not yet suggest that these patients are at increased risk of having an adverse outcome of COVID-19. However, as yet no systematic analyses have been performed to inform physicians whether JAKi therapy may be continued or should be stopped prior to or during infection. Regardless, since these patients may be at an increased risk, primary prevention should be stressed with rigorous application of recommended public health and behavioural measures applied, including physical distancing, wearing masks and hygienic measures as recommended by most governments worldwide. Prophylactic discontinuation of an effective anti-inflammatory or immune modulatory therapy is not recommended at this time.134–136 However, if therapy has been temporarily ceased in IMID patients with proven COVID-19 infection, when to safely recommence therapy is also not known for patients that have recovered. It is suggested that when oropharyngeal PCR swabs are negative virus shed after a further 7 days may be non-viable.137
To better understand the consequences of COVID-19 on IMID patients with and without specific therapies, it is critical to enter patients infected with SARS-CoV2 into relevant registries and several exist.138–140 Of note, JAKi has been suggested to be potentially beneficial against COVID-19, particularly in the context of the cytokine release syndrome like hyperinflammation which occurs in a small subset of patients, and several trials are currently ongoing to learn whether this approach has positive, negative or neutral effects.141–144 A rationale for the potential efficacy of baricitinib has been recently provided by Stebbing et al and based on predicted interference with viral trafficking.145 However, answers to the clinical validity of these hypotheses must come from observational studies as well as properly performed clinical trials.
In summary, JAKi are a new class of agents for the treatment of a variety of IMIDs with efficacy in many indications that is at least as good as that of bDMARDs and with an acceptable safety profile. Given their non-protein nature, antidrug antibodies and thus a potential secondary loss of efficacy would not occur. It is anticipated that based on these qualities and on the fact that they can be taken by the oral route their use will significantly increase over time. The presented consensus statement may be particularly helpful to those prescribing these drugs who aim to achieve the most appropriate and optimal use of these therapies.
Footnotes
Handling editor Dimitrios T Boumpas
Contributors All authors participated in the disucssions and in the work on the manuscript and its revision.
Funding This research was funded by AbbVie and Eli Lilly and Company.
Disclaimer The views expressed in this paper are those of the authors and not necessarily those of any of the individuals' employers or funders. Readers should be aware and the authors realise that in the future new and possibly conflicting data will arise and there may be errors that were unknown or not realized at the time of deliberations and the writing of this paper, but look forward to updating the paper periodically as new data come to hand and research agenda items are addressed.
Competing interests DA has received grants from AbbVie, Novartis and Roche, consulting and lecture fees from Abbvie, Amgen, Celgene, Lilly, Medac, Merck Sharpe & Dohme, Novartis, Pfizer, Roche, Sandoz and Sanofi/Genzyme. W-HB has received a research grant from Pfizer and honoraria for advice from Abbvie, Alimirall, BMS, Celgene, Janssen, Leo, Lilly, Novartis and UCB. TD received study support and/or consulting fees by AbbVie, Celgene, Eli-Lilly, EMD/Merck, Janssen, Novartis, Pfizer, Roche, Sanofi, UCB Pharma. MD: grants from Pfizer, Abbvie, UCB, Janssen, Novartis and honoraria from Pfizer, Abbvie, UCB, Janssen, MSD, Novartis, BMS, Celgene, Biogen, Sandoz. PE: grants from AbbVie, Novartis, Samsung, Lilly and honoraria from AbbVie, Novartis, BMS, Gilead, Samsung, Lilly. XB, KG received consultancy and lecture fees from Novartis, Pfizer and Roche and investigational grants from Roche. EBL reports grants from Green pharma, Seoul, grants from Handoc, Seoul, and personal fees from Pfizer. PTN has received research grants and funding for clinical trials and honoraria for advice and lectures on behalf of Abbvie, BMS, Boehringer Ingelheim, Celgene, Eli-Lilly, Gilead, Janssen, Merck, Novartis, Pfizer, Roche, Sanofi, UCB Pharma. JEP has consulted for Amgen, AbbVie, BMS, Celltrion, Gilead, Janssen, Lilly, Merck, Novartis, Pfizer, Roche, Samsung, Sandoz, Sanofi, Teva, UCB. RMF has received research grants and funding for clinical trials and honoraria for adbice from Abbvie, Amgen, Astra-Zeneca, BMS, Celltrion, Lilly, Gilead, GSK, Janssen, Novartis, Pfizer, Regeneron, Roche, Sandoz, Sanofi, Tahio and UCB. DvdH received consulting fees AbbVie, Amgen, Astellas, AstraZeneca, BMS, Boehringer Ingelheim, Celgene, Cyxone, Daiichi, Eisai, Eli-Lilly, Galapagos, Gilead, Glaxo-Smith-Kline, Janssen, Merck, Novartis, Pfizer, Regeneron, Roche, Sanofi, Takeda, UCB Pharma and is Director of Imaging Rheumatology bv. JDI has received research funding or honoraria from Abbvie, Bristol-Myers Squibb, Eli Lilly, Gilead, Janssen, Merck, Pfizer, Roche. AK has received lecture fees from Bristol-Myers Squibb, Celgene, Merck Sharp and Dohme and Pfizer. JK received grant support for clinical research from Abbvie, BMS, Lilly, Novartis, Pfizer and consulted with Abbvie, BMS, Lilly, Novartis, Pfizer and Regeneron/Sanofi. IBM has received research funding or honoraria from Abbvie, Astra Zeneca, Celgene, GSK, Lilly, Boehringer, Pfizer, Janssen, Novartis, UCB, BMS, Sanofi. WPM has served as a consultant, and/or received honoraria and/or has received research/educational grants from AbbVie, Boehringer Ingelheim, Celgene, Eli Lilly and Company, Galapagos, Janssen, Novartis, Pfizer and UCB Pharma, and is Chief Medical Officer of CARE Arthritis Limited. MV: no conflict of interest. JSS has received research Grants for his institution from AbbVie, BMS, Lilly, MSD, Pfizer, Roche and honoraria for consultancies and/or speaking engagements: Abbvie, Amgen, Astra-Zeneca, Astro-Celltrion, BMS, Celgene, ILTOO, Janssen, Lilly, MSD, Novartis-Sandoz, Pfizer, Roche, Samsung, Sanofi, UCB. MS-M has no conflicts of interest. LST received grant support from Janssen, GSK and Pfizer and honoraria for consultations and/or speaking engagements from Janssen, Pfizer, Sanofi, Abbvie and Lilly. TT received grants from Astellas, AbbVie GK, Eisai; speaking fee from: AbbVie GK, Eli Lilly Japan, Astellas, Eisai, Gilead, Pfizer Japan; and consultancy fees from: Astellas, AbbVie GK, Gilead. YT has received speaking fees and/or honoraria from Daiichi-Sankyo, Eli Lilly, Novartis, YL Biologics, Bristol-Myers, Eisai, Chugai, Abbvie, Astellas, Pfizer, Sanofi, Asahi-kasei, GSK, Mitsubishi-Tanabe, Gilead, Janssen and has received research grants from Mitsubishi-Tanabe, Chugai, Abbvie, Takeda, UCB, Daiichi-Sankyo, Eisai. MT: Speaker for BMS, Falk Foundation, Gilead and MSD; advisory boards for Albireo, BiomX, Boehringer Ingelheim, Falk Pharma GmbH, Genfit, Gilead, Intercept, MSD, Novartis, Phenex and Regulus. He further received unrestricted research grants from Albireo, Cymabay, Falk, Gilead, Intercept, MSD and Takeda. He is also co-inventor of patents on the medical use of norUDCA (filed by the medical University of Graz as previous employer). FvdB received consultancy and/or speaker fees from Abbvie, Celgene, Eli Lilly, Galapagos, Gilead, Janssen, Merck, Novartis, Pfizer and UCB. MdW has received honoraria for consultancies and speaking through Stichting Tools from AbbVie, BMS, Celgene, Janssen, Lilly, Novartis, Pfizer, Roche. RW: honoraria for speaking engagements and advice from Galapagos, Gilead and Celltrion. KLW received grant support from BMS and Pfizer and honoraria for consultations and /or speaking engagements from Pfizer, BMS, Abbvie, Gilead, Galapagos, Lilly, GSK, UCB. RX received consultancy and/or speaker fees from Abbvie, Lilly, Pfizer, UCB, Janssen.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.