Article Text

Abstract
Objectives To evaluate the efficacy and safety of certolizumab pegol (CZP) after 24 weeks in RAPID-PsA (NCT01087788), an ongoing Phase 3 trial in patients with psoriatic arthritis (PsA).
Methods Patients were randomised 1:1:1 to placebo, 200 mg CZP every 2 weeks (Q2W) or 400 mg CZP every 4 weeks (Q4W). Patients could have had exposure to one previous tumour necrosis factor (TNF) inhibitor therapy. Primary endpoints were American College of Rheumatology 20% (ACR20) response at week 12 and modified Total Sharp Score change from baseline at week 24. Secondary endpoints included; Psoriatic Arthritis Response Criteria (PsARC) score, Health Assessment Questionnaire Disability Index (HAQ-DI), Psoriasis Area and Severity Index, Leeds Enthesitis Index, Leeds Dactylitis Index, and Modified Nail Psoriasis Severity Index.
Results Of 409 patients randomised, 368 completed 24 weeks of treatment. ACR20 response was significantly greater in CZP 200 mg Q2W and 400 mg Q4W-treated patients than placebo (58.0% and 51.9% vs 24.3% (p<0.001)) at week 12, with improvements observed by week 1. There was a statistically significant improvement in physical function from baseline, measured by HAQ-DI in CZP patients compared with placebo (−0.50 vs −0.19, p<0.001) and more patients treated with CZP 200 mg Q2W and CZP 400 mg achieved an improvement in PsARC at week 24 than placebo (78.3% and 77.0% vs 33.1% (p<0.001)). Sustained improvements were observed in psoriatic skin involvement, enthesitis, dactylitis and nail disease. Higher ACR20 response with CZP was independent of prior TNF inhibitor exposure. No new safety signals were observed.
Conclusions Rapid improvements in the signs and symptoms of PsA, including joints, skin, enthesitis, dactylitis and nail disease were observed across both CZP dosing regimens.
- Anti-TNF
- Treatment
- Psoriatic Arthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Psoriatic arthritis (PsA) is a chronic inflammatory arthritis affecting up to 30% of psoriasis patients.1 ,2 The long-term burden of disease is substantial with over half the patients developing progressive, erosive disease associated with functional impairment.3 ,4
Activated T cells and proinflammatory cytokines, particularly tumour necrosis factor (TNF), play an important role in the pathophysiology of PsA.5–7 TNF inhibitors have been demonstrated to improve the musculoskeletal and skin manifestations of PsA.8–12 Certolizumab pegol (CZP), a PEGylated Fc-free anti-TNF, is clinically effective in the treatment of rheumatoid arthritis (RA)13–15 and has been shown to be effective in the treatment of psoriasis during a Phase 2 trial.16
This is the first report of clinical efficacy and safety of CZP to week 24 from an ongoing Phase 3 trial in patients with PsA (RAPID-PsA). The RAPID-PsA (NCT01087788) trial is a 216-week, randomised, double-blind, multicentre trial which was placebo-controlled to week 24. The efficacy of CZP on psoriatic skin symptoms, enthesitis, dactylitis, and nail disease is also reported. RAPID-PsA is the first published randomised controlled trial (RCT) of a TNF inhibitor in PsA to include patients with prior TNF inhibitor exposure.
Methods
Patients
The study randomised 409 patients aged 18 years or over with a diagnosis of adult-onset PsA of at least 6 months’ duration, defined by the Classification Criteria for Psoriatic Arthritis (CASPAR) group criteria (see online supplementary table S1).17 Patients had to have active joint disease, defined as ≥3 tender joints, ≥3 swollen joints, and either erythrocyte sedimentation rate ≥28 mm/h (Westergren) or C-reactive protein (CRP) >upper limit of normal (7.9 mg/L), and have previously failed ≥1 disease-modifying antirheumatic drug (DMARD). Patients were required to have active psoriatic skin lesions or a documented history of psoriasis. Up to 40% of patients could have received a prior TNF inhibitor, with a washout period of >3 months before baseline visit (28 days in the case of etanercept).
Patients with evidence of latent or active tuberculosis (TB) (PPD>5 mm) were excluded unless prophylactic treatment of latent TB had begun ≥4 weeks prior to baseline. Other exclusion criteria included evidence of: chronic or clinically significant infections, malignancy, or demyelinating disease of the central nervous system; previous exposure to >2 biologics or >1 TNF inhibitor for the treatment of PsA or psoriasis, or primary failure of a prior TNF inhibitor according to investigator assessment; and a diagnosis of any other inflammatory arthritis. Concomitant methotrexate (MTX, up to 25 mg/week), sulfasalazine (SSZ, up to 3 g/day), or leflunomide (LEF, up to 20 mg/day) treatment, maintained at a stable dose, was allowed, but not required, if it had been initiated ≥28 days before the baseline visit. Oral corticosteroids at a stable dose ≤10 mg/day prednisone or equivalent were permitted. The use of DMARDs other than MTX, SSZ, LEF, or intra-articular corticosteroids, was prohibited within 28 days of the baseline visit and during the trial. Use of combinations of MTX, SSZ and LEF was not permitted. Concurrent use of topical, systemic, or phototherapy treatments was not permitted up to week 48 of the study.
Study design
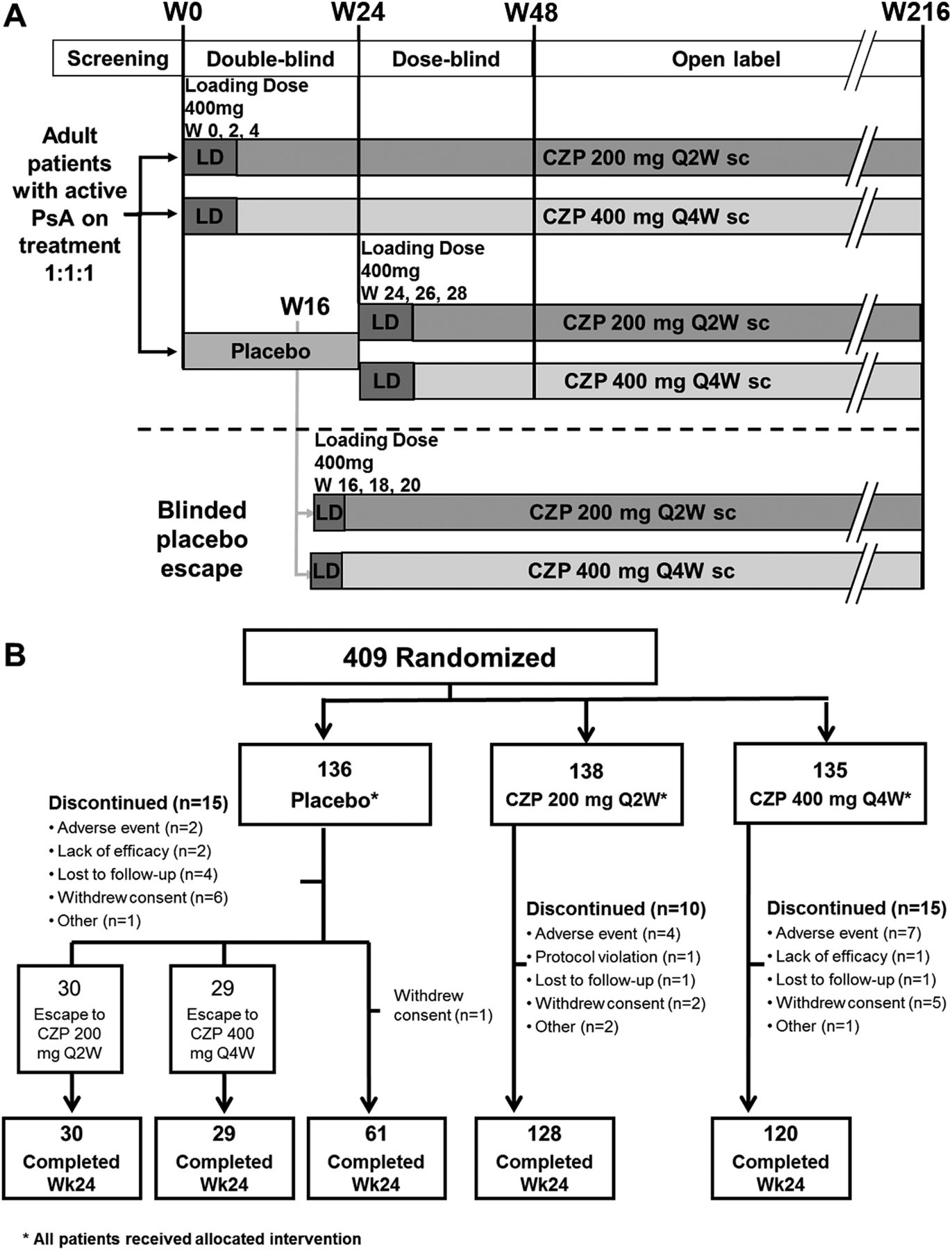
The trial is double-blind and placebo-controlled to week 24, dose-blind to week 48, and then open label to week 216 (figure 1A), and is being conducted at 92 centres in North America, Latin America, Western Europe and Central/Eastern Europe. The first patient was enrolled on 2 March 2010, and the last patient completed the 24-week period on 3 November 2011. The study was approved by the institutional review boards at participating sites and written informed consent was obtained from all patients before any protocol-specific procedures were performed.

Study design and patient disposition. *All patients received trial medication; CZP: certolizumab pegol; Q2W: every 2 weeks; Q4W: every 4 weeks; LD: loading dose; PsA: psoriatic arthritis; sc: subcutaneous; Wk: week. (A) Study design of RAPID-PsA. (B) Patient disposition to week 24.
Patients were randomised 1:1:1 to placebo (0.9% saline), or 400 mg CZP at week 0, 2 and 4 loading dose followed by either 200 mg CZP every 2 weeks (Q2W) or 400 mg CZP every 4 weeks (Q4W), administered subcutaneously by investigators using a blinded prefilled syringe (the cumulative monthly dose of CZP was the same for subjects randomised to CZP). Randomisation was stratified by investigator site and prior exposure to TNF inhibitor according to an interactive voice response system. Placebo patients who failed to achieve a 10% improvement from baseline in both swollen and tender joints at weeks 14 and 16 underwent mandatory escape to active treatment in a blinded manner. These patients were re-randomised to active treatment at week 16 in a 1:1 ratio (CZP 200 mg Q2W: CZP 400 mg Q4W); receiving loading doses at weeks 16, 18 and 20. All CZP patients continued to receive the initially assigned dose.
Study procedures and evaluations
The clinical primary endpoint was the American College of Rheumatology 20% (ACR20) response18 at week 12 using the Health Assessment Questionnaire Disability Index (HAQ-DI)19 and CRP levels.
The radiographic primary endpoint of the trial was change from baseline to week 24 in the van der Heijde modified Total Sharp Score (mTSS), and is presented separately.20 ,21
Key secondary endpoints were ACR20 at week 24, change from baseline in HAQ-DI at week 24, and 75% reduction in the Psoriasis Area and Severity Index (PASI75) in patients with at least 3% body surface area (BSA) psoriatic skin involvement at baseline at week 24.22
Other secondary endpoints included ACR50 and ACR70 responses; ACR20 responses at all visits; PASI90 response at week 24; ACR20 response rates based on stratification by baseline prior TNF inhibitor exposure, and concomitant and prior DMARD use; change from baseline in the Leeds Enthesitis Index (LEI)23 in patients with LEI ≥1 (enthesitis) at baseline and the Leeds Dactylitis Index (LDI)23 in patients with dactylitis (≥1 dactylitis digit with a circumference ≥10% larger compared with the contralateral digit) at baseline. Other outcomes included the Psoriatic Arthritis Response Criteria (PsARC)24 score at week 24, PASI50 at week 24, and in patients with nail disease at baseline, the Modified Nail Psoriasis Severity Index (mNAPSI, assessing only the most affected nail at baseline).25
Minimal disease activity (MDA) as defined and validated by Coates et al26–28 at week 24 was analysed posthoc, as were PsARC by concomitant DMARD use, ACR50 and ACR70 response by prior TNF inhibitor exposure, and PASI response by baseline PASI score.
Safety evaluations included adverse events (AEs) and routine laboratory analyses performed at every study visit through week 24.
Statistical analysis
A hierarchical test procedure was applied to protect the overall significance level for the multiplicity of dose groups and endpoints. The predefined order of hypotheses testing, each at a two-sided α=0.05 versus placebo, was: (1) ACR20 response at week 12 (CZP 200 mg Q2W or 400 mg Q4W), (2) ACR20 response at week 24 (CZP 200 mg Q2W or 400 mg Q4W), (3) HAQ-DI change from baseline at week 24 (CZP combined), (4) mTSS change from baseline at week 24 (CZP combined), (5) PASI75 at week 24 (CZP combined), (6) mTSS change from baseline at week 48 (CZP combined).
Analyses were conducted in the randomised set (RS), which included all patients randomised to the study. A sample size of 130 patients for each treatment group had a power of 99% to detect a statistically significant difference in ACR20 between placebo and CZP dose regimens (assuming a 25% difference), and a power of 95% to detect a difference in mTSS change from baseline between placebo and the combined CZP group (assuming a difference of 1 and SD of 2.4, based on radiographic difference observed in previous trials of TNF inhibitors in PsA29).
ACR responses were assessed using a standard 2-sided Wald asymptotic test with an α=0.05. The difference between placebo and combined CZP dose groups for PASI75, PASI90, PsARC and MDA response was similarly determined. CIs for ACR, PASI75 and PASI90 responses were calculated using the Wilson method30 posthoc. Non-responder imputation was used for ACR, PASI, PsARC and MDA responses, in which patients who discontinued from the study, or had missing data, were considered non-responders. Patients who escaped placebo treatment at week 16 were considered placebo non-responders from week 16 onwards. CZP patients eligible for escape continued to receive CZP and were not assumed to be non-responders from week 16.
Treatment comparisons for the combined CZP dose groups versus placebo for change from baseline HAQ-DI, LEI, and LDI were performed using analysis of covariance models for each outcome, with treatment, region and prior TNF inhibitor exposure as model factors and baseline score as covariate. Last observation carried forward (LOCF) imputation methodology was used for missing HAQ-DI, LEI, LDI and mNAPSI data.
Safety analyses included all patients who received at least one dose of study medication. Number of patients experiencing each AE is presented for each treatment group.
Results
Patient disposition and baseline characteristics
There were 409 patients randomised who received at least one dose of study medication, 368 (90.0%) patients completed the 24-week phase (figure 1B). Reasons for discontinuation from each treatment group are presented in figure 1B. A total of 59 (43.4%) placebo patients were re-randomised to CZP treatment at week 16. Additionally, 18 (13.0%) CZP 200 mg Q2W and 21 (15.6%) CZP 400 mg Q4W patients fulfilled escape criteria, but continued randomised treatment with the same dose regimen as per protocol.
The demographic and baseline characteristics of the treatment groups were generally well balanced (table 1). Over half (64.3%) the patients randomised had enthesitis at baseline, and 26.4% had confirmed dactylitis. Most patients (61.6%) had at least 3% BSA skin involvement, and patient's mean baseline HAQ-DI (1.3±0.7) indicated impaired physical function. Baseline PASI was over 50 for 5 patients (50–55 for 4 patients, 72 for 1 patient). Concomitant DMARDs were used by 70.2% of patients at baseline through week 24. A total of 19.1% of placebo and 19.8% of CZP (combined dose) patients had received prior TNF inhibitor treatment. Reasons for prior TNF inhibitor withdrawal included secondary failure, AEs, and other reasons including financial and supply problems.
Baseline demographics and disease severity characteristics, by treatment group*
Efficacy
Clinical response
At week 12, significantly more patients in the CZP 200 mg Q2W and CZP 400 mg Q4W groups achieved an ACR20 response (primary clinical endpoint) compared with patients receiving placebo (58.0% and 51.9% vs 24.3%, p<0.001, figure 2A). A clinically significant difference in ACR20 response between both CZP treatment groups and placebo was observed as early as week 1 (p<0.001) and continued through week 24 (see online supplementary figure S1). Substantial differences in CZP groups compared with placebo were seen from week 4 in ACR50 and ACR70 (figure 2B). Differences continued to week 24 (ACR50: 42.1% vs 12.5%, ACR70: 26.0% vs 4.4%, for CZP combined groups and placebo, respectively, p<0.001). Online supplementary table S2 shows the ACR core set measures. Higher placebo response rates were observed for ACR20 responses at week 12 in Latin America compared with Europe, North America (see online supplementary table S3).

Effects of certolizumab pegol treatment on PsA disease activity and psoriasis. CZP: certolizumab pegol; Q2W: every 2 weeks; Q4W: every 4 weeks; PBO: placebo; PsA: psoriatic arthritis. (A) Percentages of patients achieving a response according to the American College of Rheumatology Criteria for 20% improvement (ACR20), 50% improvement (ACR50), and 70% improvement (ACR70) at week 12 and week 24, by treatment group. *p<0.001; †p=0.003 versus placebo. (B) Percentages of patients achieving an ACR20, ACR50 and ACR70 response over time, by treatment group. *p<0.001; †p=0.005 versus placebo. (C) Percentages of patients with 3% body surface area (BSA) psoriasis at baseline achieving a Psoriasis Area and Severity Index 50% improvement (PASI50), 75% improvement (PASI75) and 90% improvement (PASI90) at week 12 and week 24. No statistical testing was conducted on PASI50 response. *Nominal p value <0.005 versus placebo. (D) Percentages of patients with 3% BSA psoriasis at baseline achieving a PASI50, PASI75 and PASI90 response over time, by treatment group. No statistical analysis was conducted on PASI50 response. *Nominal p value <0.001; †p value=0.016 versus placebo.
Treatment with CZP resulted in statistically and clinically significant improvements in physical function compared with placebo, measured by mean change in HAQ-DI at week 24 (combined CZP groups: −0.50 vs placebo: −0.19, p<0.001); the difference between CZP and placebo-treated patients was seen by week 2 (−0.23 vs −0.13, p=0.005).
In patients with ≥3% BSA psoriasis involvement at baseline, PASI50, PASI75 and PASI90 response occurred more frequently in the CZP groups at weeks 12 and 24 compared with placebo (figure 2C). At week 24, 62.2% and 60.5% of patients treated with CZP 200 mg Q2W and CZP 400 mg Q4W, respectively, achieved a PASI75 response, compared with 15.1% in the placebo group (p<0.001); a substantial difference was seen at week 2 (p<0.05) (figure 2D). PASI90 response rates were higher in CZP groups compared with placebo from week 12 through week 24 (p<0.05). PASI50 rates favoured CZP, although no statistical testing was performed. At week 24, higher PASI75 response rates were observed in patients with a PASI score ≥10 at baseline compared with those with PASI <10 (81.1% and 73.5% vs 14.3% compared with 49.1% and 50.0% vs 15.5% in CZP 200 mg Q2W and CZP 400 mg Q4W vs placebo).
In the RS, a clinically relevant difference in PsARC response rates between patients in the CZP 200 mg Q2W and 400 mg Q4W groups versus placebo was observed by week 1 (33.3% and 35.6% vs 14.0%, respectively, p<0.001) and was maintained through week 24 (78.3% and 77.0% vs 33.1%, p<0.001).
At week 24, MDA was achieved in 33.3% and 34.1% of CZP 200 mg Q2W and CZP 400 mg Q4W patient groups, respectively, compared with 5.9% of placebo patients (p<0.001, see online supplementary figure S2).
In patients with baseline enthesitis, LEI change from baseline at week 24 favoured the CZP groups, with a mean change of −2.0 in the CZP 200 mg Q2W arm (p<0.001) and −1.8 in the CZP 400 mg Q4W arm (p=0.003) compared with −1.1 in placebo-treated patients (see online supplementary table S4). For patients with baseline dactylitis, mean change from baseline in LDI was also lower at 24 weeks in the CZP 200 mg Q2W and CZP 400 mg Q4W versus placebo (−40.7 (p=0.002) and −53.5 (p<0.001) vs −22.0) (see online supplementary table S4). For patients with baseline nail disease (73.3%), mNAPSI change from baseline at week 24 was −1.6 with CZP 200 mg Q2W and −2.0 with CZP 400 mg Q4W versus −1.1 with placebo (p=0.003 and p<0.001, respectively).
Patients with prior TNF inhibitor exposure
Baseline characteristics of patients with and without prior TNF inhibitor exposure, a stratification factor, were similar (see online supplementary table S5). Improvements in ACR responses at week 12 and week 24 in CZP patients compared with placebo patients were observed irrespective of prior TNF inhibitor exposure (figure 3A). ACR20 response difference from placebo was seen by week 1 in patients with prior TNF inhibitor experience (p<0.05) and those without (p<0.001, figure 3B).

{kind=link}
{kind=link}
{kind=link}
Effect of CZP in patients with and without prior TNF inhibitor exposure at week 24 in terms of ACR response and the kinetics of ACR response. CZP, Certolizumab pegol; Q2W, every 2 weeks; Q4W, every 4 weeks; PBO, placebo; TNF, tumour necrosis factor. (A) Percentages of patients with and without prior TNF inhibitor exposure achieving a response according to the American College of Rheumatology Criteria for 20% improvement (ACR20), 50% improvement (ACR50), and 70% improvement (ACR70) at week 24, by treatment group. (B) Percentages of patients with prior TNF inhibitor exposure achieving an ACR20, ACR50, and ACR70 response over time, by treatment group. (C) Percentages of patients without prior TNF inhibitor exposure achieving an ACR20, ACR50 and ACR70 response over time, by treatment group; *p<0.001; †p<0.05 versus placebo.
Effect of concomitant DMARD use
Overall, 199 of 273 (73.0%) CZP patients and 88 of 136 (64.7%) placebo patients were receiving concomitant DMARD at a stable dose through to week 24. Baseline concomitant DMARD status was not a stratification factor in randomisation, however, patients with and without concomitant DMARD had similar baseline characteristics (see online supplementary table S6). MTX was the most common concomitant DMARD, with similar use across treatment groups (table 1). Concomitant DMARD use did not appear to affect response to CZP (combined dose) with similar ACR20 (56.8 vs 50.0) and PsARC (68.3 vs 73.0) responses at week 12, for patients with and without concomitant DMARD use respectively (table 2). Improved ACR20 responder rates for patients with prior use of either 1 DMARD or ≥2 DMARDs were observed for all CZP treatment groups compared with placebo at weeks 12 and 24 (p<0.001) (see online supplementary table S7).
ACR20 and PsARC responder rate at week 12 by concomitant use of DMARDs at baseline
Safety
Rates of AEs, serious AEs, and infections were similar between treatment groups through week 24 (table 3). The most common non-infectious AEs were diarrhoea (3.6% CZP vs 2.9% placebo) and headache (3.6% CZP vs 1.5% placebo) (see online supplementary table S5). The most common infectious AEs were nasopharyngitis (8.7% CZP vs 7.4% placebo) and upper respiratory tract infection (7.8% CZP vs 5.1% placebo). Most AEs were mild to moderate in severity. No individual type of serious AE occurred in more than one patient. Increases in liver enzymes were reported more frequently in CZP patients (see online supplementary table S8).
Adverse events during the 24 week, placebo-controlled, double-blind phase, by treatment group
Two deaths occurred during the first 24 weeks; one myocardial infarct in the CZP 200 mg Q2W group, and one sudden death of unknown cause in the CZP 400 mg Q4W group. Both deaths were considered unrelated to study medication by investigators. One patient receiving 400 mg CZP reported a non-invasive cervical carcinoma (stage 0). No congestive heart failure, serious injection site reactions, TB cases or opportunistic infections were reported.
Discussion
In the RAPID-PsA trial, CZP effectively improved the signs and symptoms of PsA with rapid onset of action (differences in clinical measures by week 1 (p<0.001)) and a statistically significant difference in ACR20 at week 12 (the primary clinical endpoint), which was maintained up to week 24. Relevant improvements were also observed for ACR50 and ACR70. Statistically significant, and clinically relevant improvement in physical function, measured as a reduction in HAQ-DI scores, was demonstrated in the CZP groups, with response seen by week 2 and maintained up to week 24. The CZP loading dose may contribute to the observed fast response within the first 4 weeks of treatment. CZP was effective with respect to improvements in the arthritis and physical function and also improved dactylitis and enthesitis, and demonstrated rapid improvements in skin involvement, measured by the PASI response. At week 24, improvements in nail disease were also observed.
The improvements in the signs and symptoms of PsA with CZP were comparable with other Phase 3 TNF inhibitor studies in PsA.9 ,12 ,31
The placebo-controlled phase of RAPID-PsA was not designed to demonstrate equivalence between the two CZP dosing schedules; however, despite differences at specific time points, kinetics curves over time did not demonstrate any clinically relevant differences between the 200 mg Q2W and 400 mg Q4W doses of CZP for any of the reported outcomes, suggesting dosing flexibility.
The treatment options for PsA are limited, with conventional DMARDs used for RA shown to have limited benefit in PsA.32 Previous trials of TNF inhibitors in PsA have demonstrated the benefit of this class of treatment,9 ,10 ,12 significantly changing the treatment paradigm in PsA; depending on local guidelines, it is now common to cycle through TNF inhibitors if therapy with DMARDs has failed, as few other treatment choices are available.33 Recent studies of ustekinumab (an interleukin 12/23 inhibitor) have also demonstrated reduced efficacy in PsA patients with prior exposure to TNF inhibitors.34
Prior TNF inhibitor exposure was a stratification factor of RAPID-PsA, and clinical results suggest a similar level of response for patients treated with CZP, irrespective of prior TNF inhibitor exposure. As patients with primary failure to previous TNF inhibitor therapy were excluded from the trial, further investigation is necessary to determine treatment efficacy in this patient population. Although not a stratification factor, patients with and without concomitant DMARDs at baseline had similar baseline characteristics and experienced similar benefits from CZP; these results are similar to those observed with other TNF inhibitors.12 ,31
Approximately 90% of patients completed 24 weeks of therapy. The rate of serious infectious events was similar in this population to what has previously been reported in patients with RA treated with CZP. There was an increased incidence of liver test elevation, generally seen in patients treated in combination with DMARDs (data not shown). Two deaths were reported during the 24-week double-blind phase of this study; the causes were what would be expected in this population of patients, and both were considered to be unrelated to study medication by the investigators.
The limitations of this report include that it is the 24-week results of RAPID-PsA, and that the results from the ongoing long-term study will be needed to confirm the sustainability of these responses. Further, the relatively low average baseline PASI score in the PsA population may explain an apparent inconsistency between the results of this trial and those previously published for CZP in moderate to severe psoriasis, as patients with PASI <10 may have had limited scope for improvements in skin involvement.16 The unusually high baseline PASI scores noted in five patients (range 50–72), if not correct, may have been due to investigator or recording error. Regional variation in ACR20 response at week 12 in the placebo group may also limit the comparison of these results with previous trials in PsA; placebo rates in South America were substantially higher than rates observed in the other geographic areas and older trials which did not include South American patients. Thus, cross-treatment comparisons may underestimate the incremental efficacy of CZP. This variation had less impact on ACR50, ACR70 and ACR20 at other time points. The use of LOCF imputation for continuous variables also leads to potential bias, particularly for patients who withdraw early from the study.
In summary, the results of RAPID-PsA confirm that treatment with CZP is clinically effective in patients with PsA up to 24 weeks, and that treatment with CZP resulted in rapid improvement in clinical signs and symptoms of PsA, including arthritis, enthesitis, dactylitis and skin involvement. Treatment with CZP in this population revealed no new safety signals from those seen in the RA trials.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Handling editor Tore K Kvien
-
Correction notice This article has been corrected twice since it was published Online First. The length of the study has been amended from 158 weeks to 216 weeks, and figure 2 has been corrected.
-
Acknowledgements The authors acknowledge Marine Champsaur, UCB Pharma, Brussels, Belgium for publication coordination and Costello Medical Consulting, UK, for writing and editorial assistance.
-
Funding The RAPID-PsA study was funded by UCB Pharma. Writing and editorial assistance was funded by UCB Pharma.
-
Competing interests UCB Pharma sponsored the trial and was responsible for data collection and analysis. The authors and the sponsor designed the study, interpreted the data, prepared the manuscript, and decided to publish. PM received grant support and/or honoraria for consultations or speaking engagements from Abbott, Amgen, Biogen Idec, Bristol Myers Squibb, Celgene, Crescendo, Forest, Genentech, Janssen, Lilly, Merck, Novartis, Pfizer, UCB Pharma. RF received grants from Genetech, Roche, Abbott, Amgen, UCB Pharma, Pfizer, Bristol Myers Squibb, Lilly, Sanofi Aventis, Lexicon, MSD, Novartis, BiogenIdec, Astellas, Astra-Zeneca, Jansen and consultant fees from Roche, Abbott, Amgen, UCB Pharma, Pfizer, BMS, Lilly, Sanofi Aventis, Lexicon, Novartis, Astellas, Astra-Zeneca, Jansen, HGS. AD received research grants and consulting fees from UCB Pharma, Abbott, and research grants from Amgen, Janssen, Novartis. JW received consultant fees from UCB Pharma. MK received non restricted grants from, Abbott, Amgen and Pfizer. DK, FW, CS, BH and TA are shareholders and employees of UCB Pharma. DvdH received consulting fees and/or research grants from AbbVie, Amgen, AstraZeneca, BMS, Centocor, Chugai, Daiichi, Eli-Lilly, GSK, Janssen Biologics, Merck, Novartis, Novo-Nordisk, Otsuka, Pfizer, Roche, Sanofi-Aventis, Schering-Plough, UCB Pharma, Vertex and is the director of Imaging Rheumatology bv.
-
Ethics approval National and Regional Ethics Committee or Institutional Review Board (Global Study).
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Clinical and epidemiological research
- Clinical and epidemiological research