Article Text

Abstract
Objectives To identify novel DNA methylation sites significant for rheumatoid arthritis (RA) and comprehensively understand their underlying pathological mechanism.
Methods We performed (1) genome-wide DNA methylation and mRNA expression profiling in peripheral blood mononuclear cells from RA patients and health controls; (2) correlation analysis and causal inference tests for DNA methylation and mRNA expression data; (3) differential methylation genes regulatory network construction; (4) validation tests of 10 differential methylation positions (DMPs) of interest and corresponding gene expressions; (5) correlation between PARP9 methylation and its mRNA expression level in Jurkat cells and T cells from patients with RA; (6) testing the pathological functions of PARP9 in Jurkat cells.
Results A total of 1046 DNA methylation positions were associated with RA. The identified DMPs have regulatory effects on mRNA expressions. Causal inference tests identified six DNA methylation–mRNA–RA regulatory chains (eg, cg00959259-PARP9-RA). The identified DMPs and genes formed an interferon-inducible gene interaction network (eg, MX1, IFI44L, DTX3L and PARP9). Key DMPs and corresponding genes were validated their differences in additional samples. Methylation of PARP9 was correlated with mRNA level in Jurkat cells and T lymphocytes isolated from patients with RA. The PARP9 gene exerted significant effects on Jurkat cells (eg, cell cycle, cell proliferation, cell activation and expression of inflammatory factor IL-2).
Conclusions This multistage study identified an interferon-inducible gene interaction network associated with RA and highlighted the importance of PARP9 gene in RA pathogenesis. The results enhanced our understanding of the important role of DNA methylation in pathology of RA.
- rheumatoid arthritis
- t cells
- autoimmune diseases
Statistics from Altmetric.com
Key messages
What is already known about this subject?
DNA methylation is a frequently studied epigenetic factor that plays an important role in the pathogenesis of RA.
Most previous epigenome-wide association studies have focused on methylation level alone, and lacked the integrationof DNA methylation with mRNA expression data to fully reveal the pathophysiological contributions and the functional roles of DNA methylation in RA.
What does this study add?
An interferon-inducible gene interaction network was associated with RA by integrating DNA methylation with mRNA expression data.
The importance of PARP9 gene was highlighted in RA pathogenesis.
How might this impact on clinical practice or future developments?
As DNA methylation plays the important roles in pathogenesis of RA, it may serve as potential biomarker and provide helpful clues for developing diagnostics, classification and therapy for RA.
Rheumatoid arthritis (RA) is a complex autoimmune disease with characteristic chronic articular synovial inflammation.1 The pathogenesis of RA so far is largely unclear yet. Epigenetic factors have recently emerged as potential elements in explaining and redefining diseases. Among a variety of epigenetic regulatory mechanisms, DNA methylation is the most frequently studied factor because of relatively more mature detection technology. The best-known function of DNA methylation is to regulate nearby gene expression. DNA methylations are most commonly observed in promoter regions where they control transcription of the nearby target genes, often in a cell-type-specific manner.
Growing evidence has suggested that DNA methylation plays an important role in the pathogenesis of RA. Early candidate gene methylation studies identified aberrant methylation changes in some specific genes, such as IL6, IL10 and CXCL12 in RA (reviewed in Klein and Gay2). Further epigenome-wide association studies (EWASs) by using modern high-throughput microarray technology, such as Illumina 450K, have been performed to systematically identify methylation markers associated with RA in various cells.3–6 These EWASs for RA have identified a large number of robust and novel candidate differentially methylated genes (DMGs), which vary widely across studies. Such inconsistency may be partially due to the great difference in study design (eg, coverage of methylation microarrays, case diagnosis, sample age, control selection, definition of differential methylation) as well as cell/tissue specificity.
6 Most previous EWASs focused on methylation level alone, which may lead to incomplete interpretation of the results regarding the functional mechanism underlying the associations between the DMGs and RA. Considering that DNA methylation normally regulates nearby gene expression, integration of DNA methylation with mRNA expression data is essential to fully reveal the pathophysiological contributions and the functional roles of DNA methylation in RA.7
To identify more novel RA-associated DNA methylation sites and fully understand their underlying pathological mechanism, we generated and integrated two omics datasets (methylome and transcriptome) from the same subjects and constructed an integrative regulatory network of RA-related DMGs. We validated the differential methylation and differential expression in two additional sample sets, and performed in-depth causal inference test (CIT)8 to evaluate the potential DNA methylation–mRNA expression–RA regulatory effect. Furthermore, we explored the potential pathogenic mechanisms of an important gene involved by conducting a series of molecular functional tests. The flow chart of the analytical pipeline and procedures is shown in online supplementary figure S1.
Supplemental material
Material and methods
Please see online supplementary methods for full details.
Supplemental material
Study subjects
A total of 43 subjects (RA:healthy control=25:18) at discovery stage, 52 subjects (RA:healthy control=25:27) for DNA methylation testing and 70 subjects (RA:healthy control=35:35) for gene expression testing at validation stage were recruited respectively. All patients with RA met the 1987 criteria of the American College of Rheumatology. The basic characteristics of all subjects are presented in online supplementary table S1.
Supplemental material
Results
1046 DNA methylation positions were associated with RA
We measured DNA methylation levels at 485 577 methylation sites in peripheral blood mononuclear cells (PBMCs) from 25 patients with RA and 18 healthy controls using Illumina Infinium HumanMethylation450K BeadChip. After quality control and screening procedure, 473 368 methylation positions were subject to differential analysis. In total, 1046 differentially methylated positions (DMPs) (|Δβ|>0.05 and detection p<0.05) including 574 hypermethylated and 472 hypomethylated DMPs (online supplementary data S1, online supplementary figure S2) were identified, which correctly separated most of RA cases and controls in the clustering analysis (online supplementary figure S3). According to the annotation, 730 DMPs were physically located within 598 unique genes. Functional enrichment analyses showed that the 598 genes were significantly enriched in some biological processes related to RA (online supplementary figure S4), for example, GO:0031295/T cell co-stimulation, GO:0006955/immune response, GO:0060333/interferon-gamma-mediated signalling pathway and GO:0050852/T-cell receptor signalling pathway. The enrichment was also found in Kyoto Encyclopedia of Genes and Genomes pathways that were confirmed related with RA9 (eg, hsa04672/Intestinal immune network for IgA production and hsa04514/Cell adhesion molecules) and several autoimmune diseases (eg, hsa05322/Systemic lupus erythematosus and hsa05320/Autoimmune thyroid disease) (online supplementary table S2). The above observations suggested that the DMGs play a critical role in the pathogenesis of RA.
Supplemental material
Identified DMPs have regulatory effects on mRNA expressions
We examined mRNA expressions in PBMCs from the same sample set (25 patients with RA and 18 healthy controls) by using the LncRNA&mRNA Human Gene Expression Microarray V.4.0 (CaptialBio, Beijing, China). Among the 598 DMP-located genes, only 445 genes (covering 540 DMPs) showed expression data in the mRNA expression microarray. Pearson correlation analyses showed significant associations between DNA methylation levels of 107 DMPs and mRNA expressions of 91 unique genes (p<0.05) corresponding to the DMPs (positive correlation:negative correlation=44:63). Among the 91 genes, 67 genes (covering 81 DMPs) were differentially expressed between RA cases and healthy controls (p<0.05). Subsequent in-depth analyses were focused on the 67 differentially expressed genes (DEGs, also DMGs) and the 81 corresponding DMPs. Of note, Poly(ADP-Ribose) Polymerase Family Member 9 (PARP9) had three DMPs located in 5′ UTR (Chr3: 122281975, cg00959259; Chr3: 122281939, cg08122652; and Chr3: 122281881, cg22930808), and its mRNA expression level also differed significantly between RA cases and healthy controls (p=3.83E−07) after Bonferroni correction (p value less than 0.05/445=1.12E−04) (online supplementary table S3).
Causal inference tests identified six DNA methylation–mRNA–RA regulatory chains
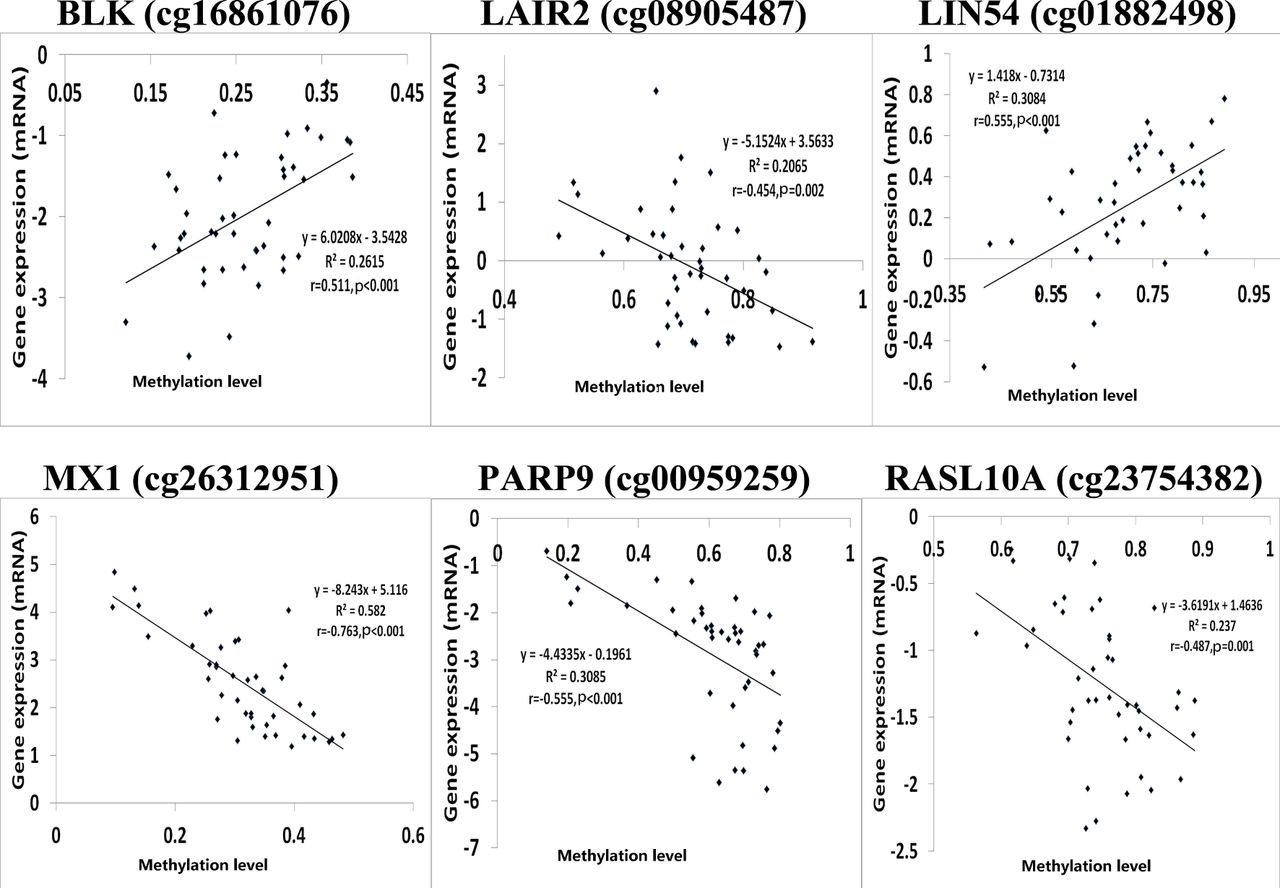
Since correlation analysis alone does not determine the causative effect, we performed in-depth CIT analyses to explore whether DNA methylation causes RA through regulating gene expression, in other words, to assess the potential regulatory chain of causal factor (DNA methylation)–mediator (mRNA)–outcome (RA). Among the above 81 identified DMPs, we selected 36 significantly correlated methylation–mRNA pairs (p<0.05 and Pearson correlation coefficients <−0.4 or >0.4) for CIT filtering. Six DMPs corresponding to six unique genes, that is, B lymphoid tyrosine kinase (BLK) (cg16861076), leucocyte-associated immunoglobulin-like receptor 2 (LAIR2) (cg08905487), Lin-54 DREAM MuvB core complex component (LIN54) (cg01882498), MX dynamin-like GTPase 1 (MX1) (cg26312951),PARP9 (cg00959259) and RAS-like family 10 member A (RASL10A) (cg23754382), were affirmed to be causative among the regulatory chains of DNA methylation–mRNA–RA (table 1, figure 1 and online supplementary table S4). Notably, PARP9 was also highlighted. Figure 1 shows the significant Pearson correlations between the six DMPs and genes identified by CIT.
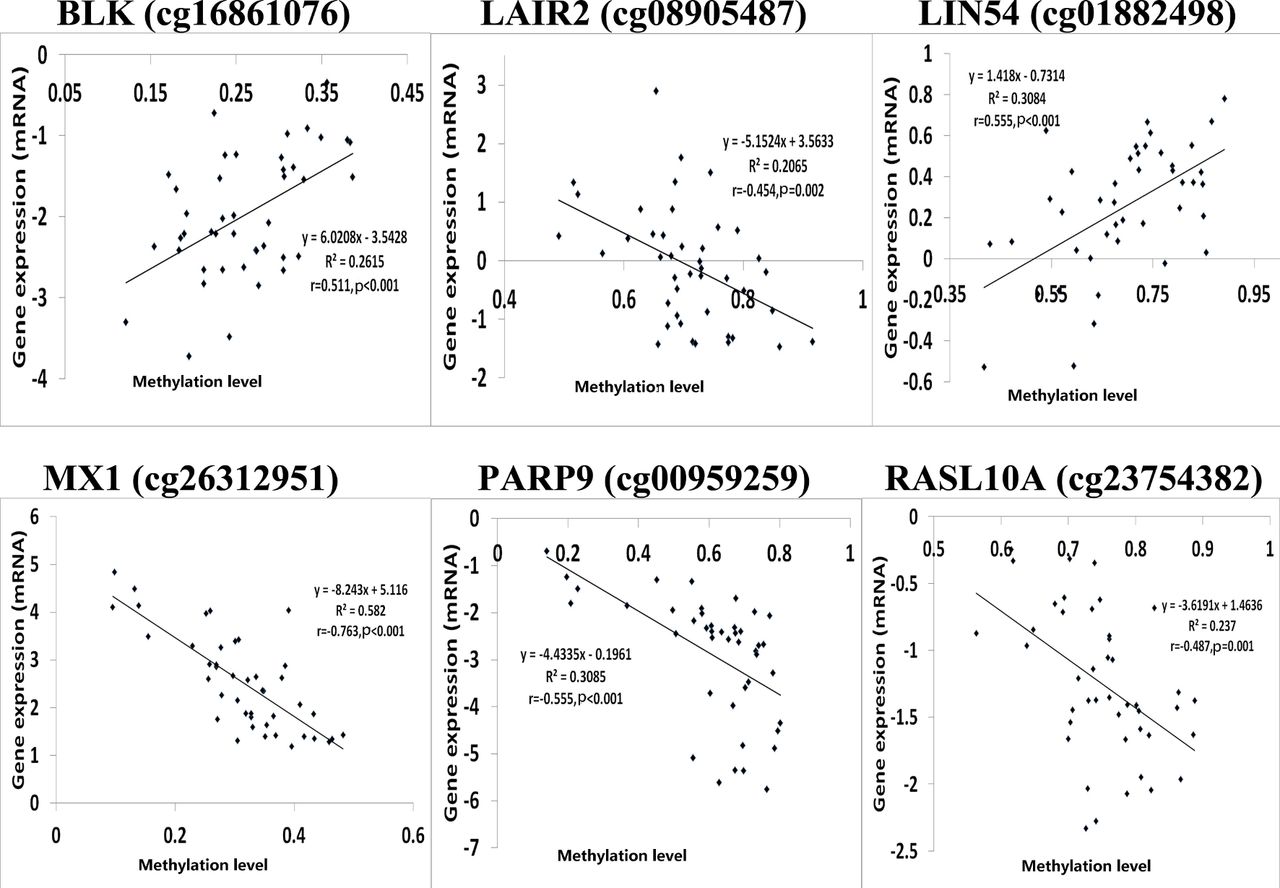
DNA methylation/mRNA correlation plots for six differential methylation positions/genes identified by causal inference test.Note: The log2 transformation was applied to the gene expression data using the Adjust Data function of Multi-experiment Viewer (MeV) software. r : Pearson correlation coefficient
Causal inference test (CIT) identified six DNA methylation–mRNA–RA regulatory chains
Identified DMPs and genes formed an interferon-inducible gene interaction network
To illustrate and better understand the regulatory effects of DMGs, we first constructed the predicted protein–protein interactions among the above 445 DMGs, 91 DMGs and 67 DMGs, respectively, using STRING,10 a database of known and predicted protein interactions. The interaction network was very complicated among the 445 DMGs (online supplementary figure S5) but much distinguishable among the 91 DMGs (online supplementary figure S6). In the network of the 91 DMGs, we found an interesting interferon-inducible gene interaction subnetwork, including MX dynamin-like GTPase 1 (MX1), ISG15 ubiquitin-like modifier (ISG15), IFN-induced protein 44-like (IFI44L), deltex E3 ubiquitin ligase 3L (DTX3L), PARP9 and signal transducer and activator of transcription 3 (STAT3). Such a network was present in the network of 67 DMGs as well (figure 2A).
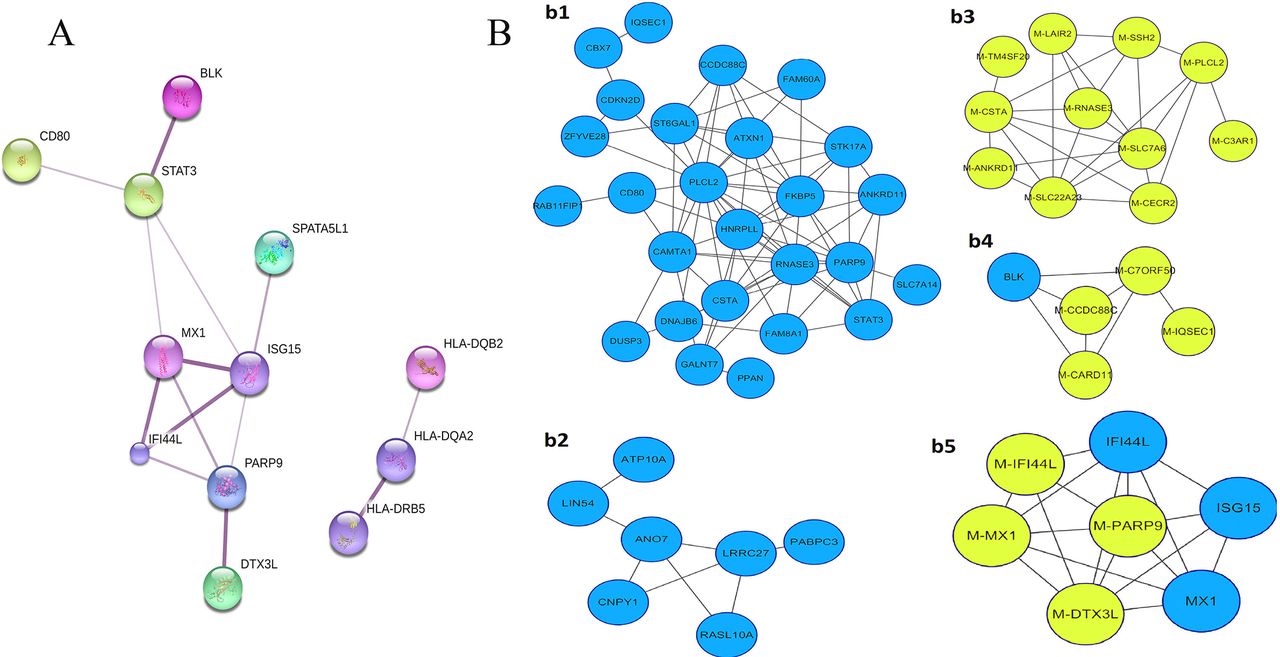
Interaction networks of differentially methylated genes (DMGs)/differentially expressed genes (DEGs). (A) Two protein–protein interaction networks constructed by STRING using the 67 DEGs. Stronger associations are represented by thicker edges. One interaction network is mainly composed of IFN-inducible genes, and the other is composed of HLA-DRB5, HLA-DQA2 and HLA-DQB2. (B) The coexpression network built by Cytoscape V.3.2.1 using β values of 81 DMPs and the mRNA expression level of 67 corresponding genes (DEGs). The yellow-green circles represent DMPs, and the blue circles represent DEGs.
{kind=link}
{kind=link}
{kind=link}
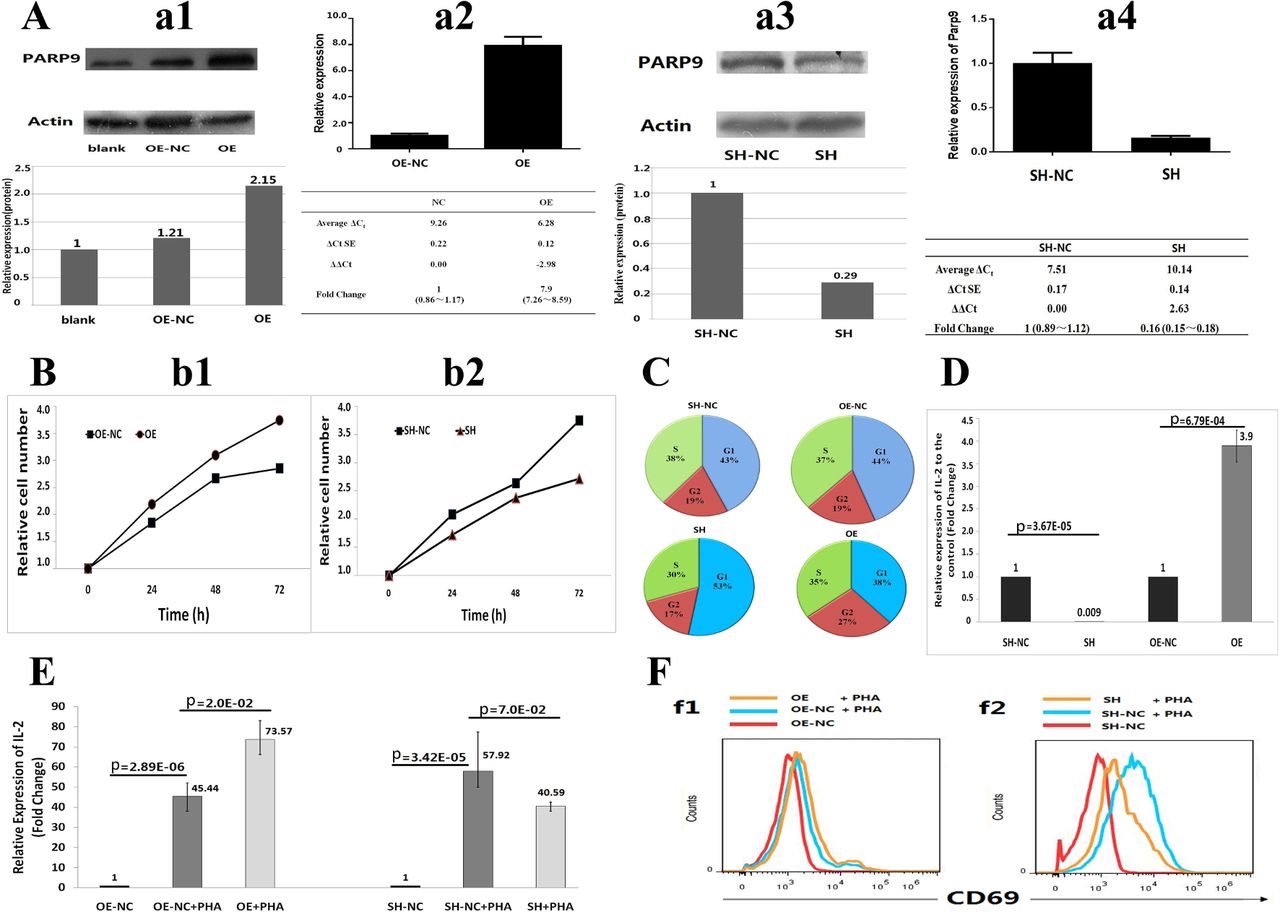
Functional tests for PARP9 in Jurkat cells in vitro. (A) Stable over-expression (OE) and down-expression (shRNA) (SH) of PARP9 in Jurkat cell line. Four pictures from left to right show western blot (WB) (A–a1) and RT-PCR (A–a2) for PARP9-OE, WB (A–a3) and RT-PCR (A–a4) for PARP9-shRNA cell lines, respectively. (B) Cell proliferation tests for PARP9-OE (B–b1) and PARP9-SH (B–b2) Jurkat cells using CCK-8 assay. Each experiment was repeated three times. (C) The percentage of stage-specific cells within the cell cycle for PARP9-OE, PARP9-OE-NC, PARP9-SH and PARP9-SH-NC Jurkat cells. Presented are mean percentages from triplicate assays. (D) The effects of PARP9 on IL-2 expression tested by quantitative RT-PCR. GAPDH was used as a reference gene in quantitative RT-PCR. (E) The effect of PARP9 on IL-2 expression during PHA stimulation. PHA, phytohaemagglutinin. (F) PARP9-regulated CD69 expression during PHA activation. Jurkat T cells were stimulated with PHA for 24 hours.The expression of CD69 in PARP9-OE and PARP9-OE-NC (F–f1), PARP9-SH and PARP9-SH-NC (F–f2) Jurkat T cells were assessed by flow cytometry. OE, PARP9 over-expression; OE-NC, negative control of PARP9 over-expression; SH, PARP9 down-expression; SH-NC, negative control of PARP9 down-expression. Each experiment was repeated three times.
We further constructed coexpression integrative networks of methylation and mRNA by using DNA methylation values (β value) of 81 DMPs and the mRNA expression data of the 67 corresponding genes into Cytoscape V.3.2.1.11 As shown in figure 2B, among the five identified subnetworks, two (figure 2B-b1,b2) contain only mRNA expression values, and PARP9 is a node gene in the network (figure 2B–b1). Two subnetworks (figure 2B–b4, b5) that contain both DNA methylation and mRNA showed the coexpression network containing MX1, PARP9, DTX3L, ISG15 and IFI44L. The results obtained through STRING and Cytoscape both pointed to an interaction network including four hub genes, MX1, IFI44L, DTX3L and PARP9.
Key DMPs and corresponding genes were validated their differences in additional samples
To verify the major results of microarray analysis, we selected 10 DMGs including PARP9, IFI44L, MX1, ISG15, family with sequence similarity 8 member A1 (FAM8A1), serine/threonine kinase 17a (STK17A), BLK, canopy FGF signalling regulator 1 (CNPY1), Chromobox 7 (CBX7) and solute carrier family 7 member 14 (SLC7A14) to validate differential DNA methylation/expression in two additional sample sets. A total of 15 DNA fragments (100–300 bp) from the 10 DMGs were analysed, and the site-specific methylation levels within each DNA fragment were quantified by targeted bisulfate sequencing. Five DMPs were validated between 25 patients with RA and 27 healthy controls, among which three (cg00959259, cg08122652, cg22930808) were located in PARP9. In addition, the quantitative RT-PCR (qRT-PCR) showed that 6 of the 10 DEGs (BLK, CBX7, IFI44L, MX1, PARP9 and STK17A) presented significant differential expressions in PBMCs between 35 patients with RA and 35 healthy controls. Four of the six genes (BLK, IFI44L, MX1 and PARP9) showed consistent regulation direction with the discovery sample (online supplementary table S5). Interestingly, most of the verified genes were involved in the interferon-inducible gene interaction network (figure 2A).
Methylation of PARP9 was correlated with mRNA level in Jurkat cells and T lymphocytes isolated from patients with RA
Since the above combined evidence highlighted the significance of DMPs in PARP9, we analysed the methylation status of the three verified DMPs and the mRNA level of PARP9 at different timepoints after 5-Aza (DNA methyltransferase inhibitor) treatment in Jurkat cells, an immortalised cell line of human T lymphocytes that are frequently used as cell model in studies of immune-related diseases.12 13 Pearson correlation analysis showed that the methylation levels of two DMPs (cg08122652 and cg22930808) were negatively correlated with mRNA expression level of PARP9 (p<0.05, r=0.356 and 0.350, respectively). The ∆CT was used in the correlation analysis in vitro. We also performed similar experiment in T cells isolated from seven patients with active RA and found a significant correlation between the methylation level of DMP (cg00959259) and PARP9 gene expression (r=0.752, p=0.019).
PARP9 gene has functional effects on Jurkat cells
We further assessed the effects of PARP9 on behaviours of Jurkat cells by constructing PARP9 over-expression (PARP9-OE) Jurkat cell line and PARP9 down-expression (shRNA) (PARP9-SH) Jurkat cell line. Comparing with its negative control, the mRNA and protein level of PARP9 was 7.9-fold and 2.15-fold upregulated, respectively, in PARP9-OE cells, and 3.5-fold and 6.3-fold downregulated, respectively, in PARP9-SH cells (figure 3A).
As expected, the PARP9-SH cells showed significantly lower proliferation rate than the negative control cells (p=0.035). Conversely, PARP9-OE cells exhibited significantly higher proliferation rate compared with the negative controls (p=0.024) (figure 3B). We also carried out cell-cycle analysis to explain the results of proliferation tests (see online supplementary figure S7) and found that reduced expression of PARP9 was associated with G1 cell-cycle arrest, as evidenced by the increased percentage of G1 and the reduced percentage of S and G2. Meanwhile, over-expression of PARP9 promoted cell proliferation, as evidenced by the reduced percentage of G1 and S and increased percentage of G2 (p<0.05) (figure 3C).
Next, we assessed the expression level of PARP9 in PARP9-OE and PARP9-SH with phytohaemagglutinin (PHA) stimulation. As shown in online supplementary figure S8, the expression level of PARP9 was significantly elevated in all the four PHA-stimulated cell lines (PARP9-OE/OE-NC and PARP9-SH/SH-NC), as compared with the unstimulated controls, respectively. Further, we determined the effect of PARP9 on inflammatory cytokine expression, such as IL-1, IL-2, IL-4, IL-6, IL-8, TNFα and IFNγ, using the qRT-PCR tests. Among the seven tested cytokines, only IL-2 presented upregulated effect in PARP9-OE and downregulated effect in PARP9-SH Jurkat cells, as compared with the negative controls, respectively. To confirm the findings, we repeated the qRT-PCR tests for IL-2 and validated the significant positive regulatory effect of PARP9 on IL-2 (p<0.05) (figure 3D). Besides, we assessed the effect of PARP9 on IL-2 expression under PHA-stimulated condition. As expected, we found that PHA could stimulate IL-2 expression in both PARP9-OE and PARP9-SH cell line. Consistent with the prior observations on unstimulated cells, the positive regulatory effects of PARP9 on IL-2 expression were also observed in PHA-stimulated PARP9-OE and PARP9-SH cell lines (figure 3E).
To further ascertain the role of PARP9 in the process of activation in Jurkat T cell, flow cytometry was employed to test the expression of antigen CD69, a commonly used early activation biomarker of T cells. We found that the CD69 expression was promoted by PHA stimulation in negative control cells. Under PHA stimulation, PARP9 over-expression slightly increased the amount of CD69 (figure 3F–f1), and PARP9 silencing evidently decreased CD69 expression (figure 3F–f2). These results indicated that PARP9 expression level could affect the degree of Jurkat T-cell activation.
Discussion
In this study, we identified an IFN regulatory gene interaction network associated with RA and ascertained the PARP9-regulated cellular mechanisms underlying RA pathogenesis. IFN has been widely used in clinic settings because of its positive effects of antiviral, inhibiting tumour cell proliferation and immune regulation. At present, the pathogenesis of autoimmune diseases including RA is poorly understood. Environmental triggers, such as virus infections, are involved in these diseases. It was hypothesised that human chronic autoimmune diseases are due to infection of autoreactive B lymphocytes by Epstein-Barr virus (EBV) and a subsequent series of T-lymphocyte reaction.14 Indeed, EBV-induced lymphoproliferative disorders were reported in about 40% of rheumatic patients.15 16 IFN was previously reported to be a potential therapy for RA that might help to diminish both joint inflammation and destruction by cytokine modulation.17 Meanwhile, a systemic upregulation of IFN type I inducible genes, the so-called IFN signature, has been demonstrated in several autoimmune diseases such as systemic lupus erythematosus (SLE), Primary Sjögren’s syndrome (pSS) and RA.18 Several studies have demonstrated that the IFN signatures in RA do have potential clinical relevance,19 and it could be served as a biomarker of preclinical RA.20 Among hundreds of IFN-inducible genes, the elevated expression of MX1 and IFI44L were most often observed in autoimmune diseases.21–25 For example, MX1 was suggested as a biomarker for IFN bioactivity in pSS,26 and the mRNA expression levels of IFI44L was increased in CD4+ T cells from patients with SLE.27 28 Here, we identified several IFN-regulated genes related to RA, including MX1, IFI44L, PARP9 and DTX3L. Hypomethylation of these IFN-regulated genes was correlated with enhanced gene expression. Our pathway analysis of DMGs also revealed IFN signalling among the top associated pathways. These findings suggest a role of type I IFN activity in the development of RA.
Epigenetic mechanisms may serve as a dynamic link between genotype, environment and phenotype. Increasing evidence suggests an epigenetic contribution to the pathogenesis of autoimmune disease.29 Considering that epigenetic alterations are potentially reversible, characterising epigenetic dysfunction in disease has the potential to identify new biomarkers and novel therapeutic targets.30 DNA methylation is the best characterised epigenetic modification in humans, and it has emerged as an important contributor to human complex diseases. Although the exact mechanisms remain to be elucidated, global hypomethylation and demethylation of IFN-regulated genes in multi-rheumatoid diseases are the main features to date.24 31 32 Loss of DNA methylation in these genes was associated with an increase in mRNA expression. Therefore, the interferon signature pathway could be featured by hypomethylation of IFN-regulatory genes.33 Moreover, a recent study revealed that significant hypomethylation of two CpG sites within IFI44L promoter was identified in patients with SLE, RA and pSS, and promising to be the first epigenetic diagnostic marker for SLE.34 Previous studies identified the significant hypomethylation of CpG site (cg06872964) within IFI44L in SLE,24 34 CpG site (cg00959259) within PARP9 in pSS24 and CpG site (cg26312951) within MX1 in pSS.24 The findings expand current knowledge of epigenetic changes of IFN type I inducible genes in autoimmune diseases. Since these epigenomic markers were also identified for RA in our study, they may serve as common sites shared by the autoimmune diseases. On the other hand, some identified DMPs may be unique to RA. For example, for the six DMPs within the CIT regulation chains (table 1), four DMPs (cg16861076, cg08905487, cg01882498, cg23754382) may be unique to RA, as they were not reported in other autoimmune diseases according to our current search (online supplementary table S6). Further research would be needed to explore whether the RA-related DMPs/DEGs identified in the present study are unique to RA or common to other autoimmune diseases.
In our study, the gene PARP9 was the most distinctive DMG in RA. Little was known about its function. In 2000, Aguiar et al 35 reported that PARP9 was a novel risk-related gene in diffuse large B-cell lymphomas that enhances cellular migration. Then, it was found that PARP9 and DTX3L are located in a head-to-head orientation, regulated by a gamma IFN-responsive bidirectional promoter, and are over-expressed in diffuse large B-cell lymphomas with a prominent inflammatory infiltrate.36 Recently, PARP9–DTX3L complex was observed with a double benefit for IFN-dependent host defense.37 As described above, our current knowledge of PRAP9 was mainly focused on its antiviral or IFN response. As a member of PARPs superfamily that has anti-inflammatory properties, DNA repair and gene transcription regulation, the functional mechanisms of PARP9 in RA remain unexplored previously. The present study demonstrated that PARP9 could influence cell cycle and cell proliferation in v itro. Reduced expression of PARP9 was associated with G1-phase arrest, and over-expression of PARP9 promoted cell proliferation in Jurkat cells. We also found that PARP9 positively regulated IL-2, a cytokine glycoprotein that stimulates a wide range of leucocytes, including T cells and natural killer cells and involved in the Th1 cytokine pathway of the immune response.38 39 Subsequent functional tests indicated that the degree of Jurkat T-cell activation was also positively regulated by PARP9 expression level. These results suggested that PARP9 could be implicated in inflammatory reactions and enhance immunoreactions of RA.
The main strength of this study is the multistage research strategy, which enables us to systematically identify both methylation sites and their target genes, and to thoroughly investigate molecular and cellular mechanisms underlying RA pathogenesis. The results enhanced our understanding of the roles of methylation in pathology of RA and provided helpful clues for developing diagnostics, classification and therapy for RA.
References
Footnotes
HZ and L-FW contributed equally.
Handling editor Josef S Smolen
Contributors HZ participated in study design, recruiting patients with RA, data analysis, molecular testing, drafted and revised the manuscript. LFW participated in molecular testing, data analysis and revised the manuscript. XBM participated in data interpretation and revised the manuscript. XL coordinated the sample collection. HT participated in sample collection, data analysis. XWZ participated in sample collection, cell culture, molecular testing and revised the manuscript. WX participated in cell culture, molecular testing, data interpretation and revised the manuscript. YFG, MJW, KQZ and JW participated in recruiting patients with RA, data analysis and revised the manuscript. YHQ and XL participated in recruiting patients with RA, gene expression data acquisition, data analysis and revised the manuscript. YHZ participated in study design and revised the manuscript. YZL and NJY participated in revising the manuscript. SFL and FYD conceived the study, participated in study design, coordinated the sample collection, revised and finalised the manuscript. All authors read and approved the final manuscript.
Funding The study was supported by Natural Science Foundation of China (81401343, 81473046,81872681, 81373010, 81502868, 31401079, 81541068), the Natural Science Foundation ofJiangsu Province (BK20150346), the Natural Science Research Project of Jiangsu Provincial Higher Education (16KJA330001), the Startup Fund from Soochow University (Q413900112,Q413900712) and a Project of the Priority Academic Program Development of Jiangsu Higher Education Institutions.
Competing interests None declared.
Patient consent Obtained.
Ethics approval The ethical committee of Soochow University.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The microarray data for methylation have been submitted to the GEO database with accession number GSE111942.