Article Text

Abstract
Objectives To evaluate equivalence in efficacy for rheumatoid arthritis (RA) and compare the safety of the biosimilar HD203 with innovator etanercept (ETN) plus methotrexate (MTX) (ClinicalTrials.gov NCT01270997).
Methods Patients with active RA received 25 mg HD203 or ETN subcutaneously twice-weekly with MTX for 48 weeks in a phase III, multicentre, randomised, double-blind, parallel-group design. The primary end point was the proportion of patients achieving the American College of Rheumatology 20% response (ACR20) at week 24 for per-protocol study completer set (PPS). Secondary end points included ACR response criteria, ACRn, European League against Rheumatism (EULAR) response, change in Disease Activity Score 28 (DAS28), patient-reported outcomes, safety and immunogenicity.
Results Of the 294 randomised patients (HD203, n=147; ETN, n=147), 233 comprised the 24-week PPS (n=115 and 118, respectively). ACR20 at week 24 was achieved by 83.48% and 81.36% of PPS patients, respectively, demonstrating equivalent efficacy within predefined margins of ±20% (treatment difference 2.12%, 95% CI −7.65% to 11.89%). Outcomes for secondary end points were consistent with the primary efficacy findings. Groups were comparable for overall incidences of treatment-emergent (all-causality) adverse events (AEs) (HD203 113 (76.9%) vs ETN 114 (78.1%) (p=0.804)), adverse drug reactions, serious AEs and discontinuations due to AEs. Few patients (HD203, n=8; ETN, n=3) tested positive for anti-drug antibodies.
Conclusion The study met the primary objective of demonstrating equivalent efficacy of HD203 and ETN. HD203 was well tolerated, with safety comparable with ETN in this population of patients with RA.
Trial registration number NCT01270997; Results.
- Rheumatoid Arthritis
- Anti-TNF
- DMARDs (biologic)
Statistics from Altmetric.com
Introduction
Etanercept is a recombinant dimeric fusion protein consisting of the two extracellular domains of the tumour necrosis factor receptor linked to the Fc portion of human immunoglobulin G1 (IgG1).1–4 When used in combination with methotrexate (MTX), innovator etanercept (ETN) has demonstrated clinical efficacy in reducing the signs and symptoms of rheumatoid arthritis (RA) in a number of randomised, controlled studies.5–10 ETN is indicated for the treatment of patients with moderate-to-severe RA who have failed or are intolerant to MTX, as well as psoriatic arthritis, ankylosing spondylitis, plaque psoriasis and juvenile idiopathic arthritis. In the European Union, ETN is also indicated for paediatric plaque psoriasis and non-radiographic axial spondyloarthritis.11 ,12
A ‘biosimilar’ is a biological medicine that is similar to another biological medicine that has already been authorised for use.13 ,14 The similarity of potential biosimilars is established as per guidelines, such as those issued by the European Medicines Agency, the Food and Drugs Administration and the WHO.15–19 Regulatory requirements for biosimilars are different from those for generic drugs and for innovative biologic agents.20 The similarity of the biosimilar to the reference product is defined in terms of biological and physicochemical characteristics (equivalent structure and pharmacokinetics (PKs)), efficacy, safety and immunogenicity.15–19
Marketed biosimilars approved by regulatory authorities have the potential to reduce costs for patients and healthcare systems, and increase the access to treatment without compromising patient outcomes.21–23 Indeed, the high cost of biologics24 often limits the access of some patients to these therapies despite their proven clinical benefit.13 ,25 ,26
HD203 is a biosimilar of ETN developed by Hanwha Chemical Biologics (Seoul, Republic of Korea). In preclinical in vitro and non-clinical analyses, HD203 has been shown to be structurally and functionally comparable with ETN, and to have similar anti-inflammatory activity and PKs (unpublished data, Hanwha Chemical Co.). In addition, a phase I study in healthy volunteers demonstrated equivalent PK profiles for HD203 compared with ETN. Tolerability of HD203 was also comparable with that of ETN.27
As a result of these initial findings, a phase III, multicentre, randomised, double-blind, active-controlled, parallel-group trial was initiated to evaluate the equivalence in efficacy and comparability of safety of HD203 25 mg and ETN 25 mg in combination with MTX in patients with Rheumatoid Arthritis (HERA). The primary objective of the study was to establish the therapeutic equivalence of HD203 and ETN in patients with RA, using prespecified equivalence criteria based on American College of Rheumatology (ACR) response rates.
Methods
Study design
This phase III, multicentre, randomised, active-controlled, parallel-group study was conducted at 37 study sites in the Republic of Korea (ClinicalTrials.gov identifier NCT01270997). Study subjects and investigators were blinded to treatment allocation (further details are provided in online supplementary appendix 1).
Supplementary Appendix
Patients were randomly assigned 1:1 to receive HD203 25 mg or ETN 25 mg (lyophilised form) administered subcutaneously (SC) twice weekly with MTX for 48 weeks. Randomisation was by an interactive web response system stratified to ensure appropriate randomisation at each study centre (see online supplementary appendix 1).
Ethical and regulatory approval and patients’ written informed consent were obtained before treatment, and the study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines.
Patients
Eligible patients were aged ≥20 years, had a diagnosis of RA according to the 1987 ACR criteria28 and had active disease, defined as ≥6 swollen joints, ≥6 tender joints, C reactive protein level ≥1.0 mg/dL or erythrocyte sedimentation rate of ≥28 mm/h. Patients were also required to be in ACR functional class I to III,29 to be positive for rheumatoid factor (RF) or anti-cyclic citrullinated protein (anti-CCP) antibody or to have bone erosions in the hands and/or feet on X-ray.
All patients had an insufficient clinical response to MTX during ≥6 months of treatment prior to screening. Patients continued to take a stable dose of MTX (7.5−25 mg/week orally, intramuscularly or SC) throughout the study. This dose was the same as the dose which had been administered for 6 weeks prior to baseline. No modifications in the MTX dose or route of administration were permitted, although a dose reduction was allowed for adverse events (AEs) within the range of 7.5−25 mg/week. Temporary suspensions of MTX administration (due to infection or surgery) were allowed for up to 14 days and for up to twice within a 6-month period. Patients receiving steroids or non-steroidal anti-inflammatory drugs were required to be maintained on the same dose of these agents for ≥4 weeks prior to baseline. Additional details on patient eligibility are provided in online supplementary appendix 1.28 ,29
Study assessments
The primary end point was the proportion of patients achieving ACR20 at week 24. Secondary end points included: ACR20 at weeks 12 and 48; ACR50 and ACR70 at weeks 12, 24 and 48; ACRn at weeks 24 and 48; change in Disease Activity Score 28 (DAS28) and DAS28 remission. Additional secondary end points included European League against Rheumatism (EULAR) response and safety and immunogenicity at weeks 24 and 48 (assessed using a bridging assay with anti-Hu-IgG detection of bound antidrug antibodies (ADAs).
Patient-reported outcomes (PROs) were assessed at baseline and at weeks 12, 24 and 48, using the Health Assessment Questionnaire (HAQ; Korean language version), Short-Form health survey, Functional Assessment of Chronic Illness Therapy on Fatigue and EuroQuol-5 dimension (Korean language version) instruments.30 ,31
Safety assessments included monitoring and recording of AEs and serious AEs (SAEs). The presence of anti-HD203 antibodies in human serum was evaluated in a qualitative test using ELISA (see online supplementary appendix 1).
Statistical analysis
Therapeutic equivalence of HD203 and ETN was determined if the 95% CI of the difference in ACR20 at 24 weeks between HD203 and ETN was contained entirely within the equivalence margin of ±20%. This equivalence level represents slightly less than half of the difference between the ACR20 response rates in ETN versus comparator treatment in the first (double-blind) clinical trial of ETN combined with MTX (71% vs 27% for placebo plus MTX).6 In order to have at least 80% power to show equivalence at a two-sided α-level of 0.05 with an assumed 71% ACR20 rate in each group, and assuming a 20% withdrawal rate, it was determined that 274 patients were required to ensure 137 patients in each group. Further evaluation of the appropriateness of the equivalence margin is included in the ‘Discussion’ section and in online supplementary appendix 2.
Descriptive statistics were calculated for baseline clinical and demographic characteristics, and the two groups were compared using two-sample t tests or Wilcoxon rank-sum test for continuous variables or Pearson's χ2 test or Fisher's exact test for categorical variables. The proportion of subjects experiencing AEs or developing laboratory abnormalities in each group was compared using Fisher's exact test or the χ2 test.
Efficacy data were analysed using the per-protocol set (PPS) as the main analysis set. The PPS included all subjects who had completed the study in accordance with the protocol (for further details, see online supplementary appendix 1). Additional intention-to-treat (ITT) analyses were undertaken on the full analysis set (FAS), which included all eligible patients who received at least one dose of the study medication and had at least one post-baseline assessment of efficacy. Treatment comparison for continuous variables such as DAS28, Simplified Disease Activity Index (SDAI), Clinical Disease Activity Index (CDAI), HAQ and PROs were assessed using analysis of covariance (ANCOVA) with baseline score as covariate. The last-observation-carried-forward (LOCF) method was used to impute any missing values. The safety analysis included all patients who received at least one dose of study medication.
Results
Patient disposition and characteristics
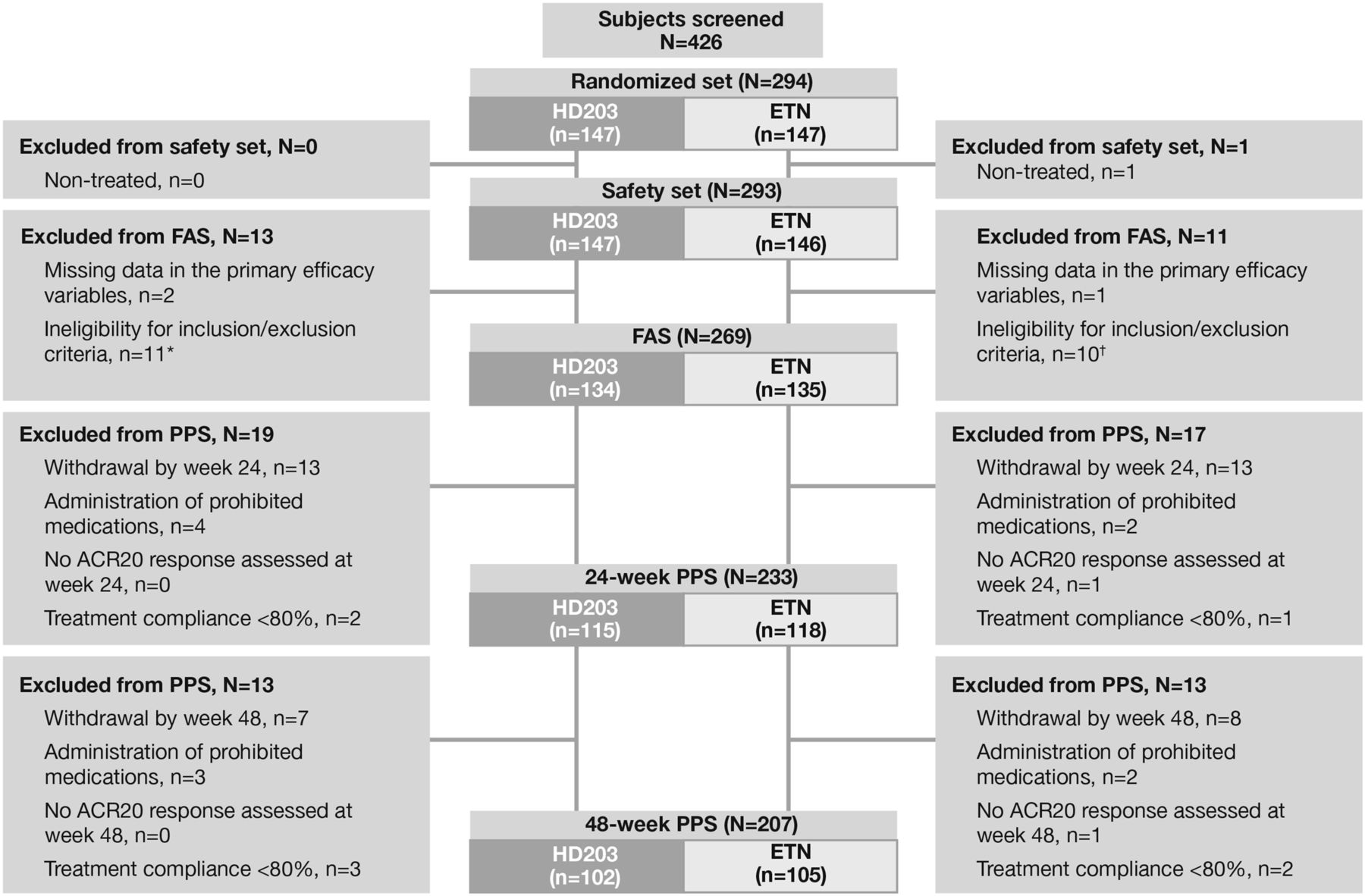
A total of 294 patients were randomised to HD203 or ETN (n=147 each) (figure 1). Totally, 240 patients completed the study (120 in both groups). The 24-week PPS included 233 patients (115 in the HD203 group and 118 in the ETN group), and the 48-week PPS included 207 patients (102 in the HD203 group and 105 in the ETN group). The FAS included 269 patients and the safety population included 293 patients. Informed consent of the first patient was obtained on 24 December 2010 and the last visit of the last patient was on 31 May 2012. The most common reason for study withdrawal before week 24 was AEs (four patients in the HD203 group and five in the ETN group), withdrawal of consent (three patients in the HD203 group and five in the ETN group) and lack of efficacy (two patients each in the HD203 and ETN groups).

Patient disposition.
All patients enrolled were ethnically Korean. Demographic and clinical characteristics were generally well balanced between the two groups (table 1 and see online supplementary table S1). However, fewer patients in the HD203 group (81.7%) than in the ETN group (91.5%) were RF-positive. The proportion of patients who tested positive for either RF or anti-CCP antibody was 93.9% in the HD203 group and 97.5% in the ETN group.
Baseline demographic and clinical characteristics (24-week PPS)
Primary end point
HD203 was similar to ETN for the proportion of patients achieving ACR20 at week 24 (primary end point): 83.48% vs 81.36% (PPS); 79.10% vs 75.56% (FAS), respectively (no significant difference) (figure 2). Equivalence in efficacy at week 24 was demonstrated within the predefined margins (95% CI of the difference in ACR20 at 24 weeks between HD203 and ETN, which was ±20%) in both the PPS (treatment difference: 2.12%; 95% CI −7.65% to 11.89%, see online supplementary table S2) and FAS (treatment difference: 3.55%; 95% CI −6.45% to 13.55%, online supplementary table S3).

(A) Proportion of patients achieving American College of Rheumatology (ACR)20 responses at week 24 in the per-protocol set (PPS) and full analysis set (FAS); (B) proportion of patients achieving ACR20 responses at week 48 (PPS); (C) proportion of patients achieving secondary endpoint ACR responses (PPS).
Secondary end points
The proportion of patients who achieved an ACR50 response was significantly higher in the HD203 group than the ETN group at week 24 and week 48 in the PPS (treatment difference (95% CI) week 24: 12.68% (0.15% to 25.20%); week 48: 13.72% (1.04% to 26.40%)) and at week 24 for the FAS (12.29% (0.45% to 24.13%)). However, no statistically significant between-group differences were observed in the proportion of patients achieving ACR20 or ACR70 responses for all time points (see figure 2 and see online supplementary tables S2 and S3). There were also no statistically significant between-group differences in ACRn either in the PPS (treatment difference (95% CI) week 24: 1.22 (−8.68 to 11.13); week 48: 4.69 (−4.81 to 14.19)) or in the FAS (see online supplementary table S3).
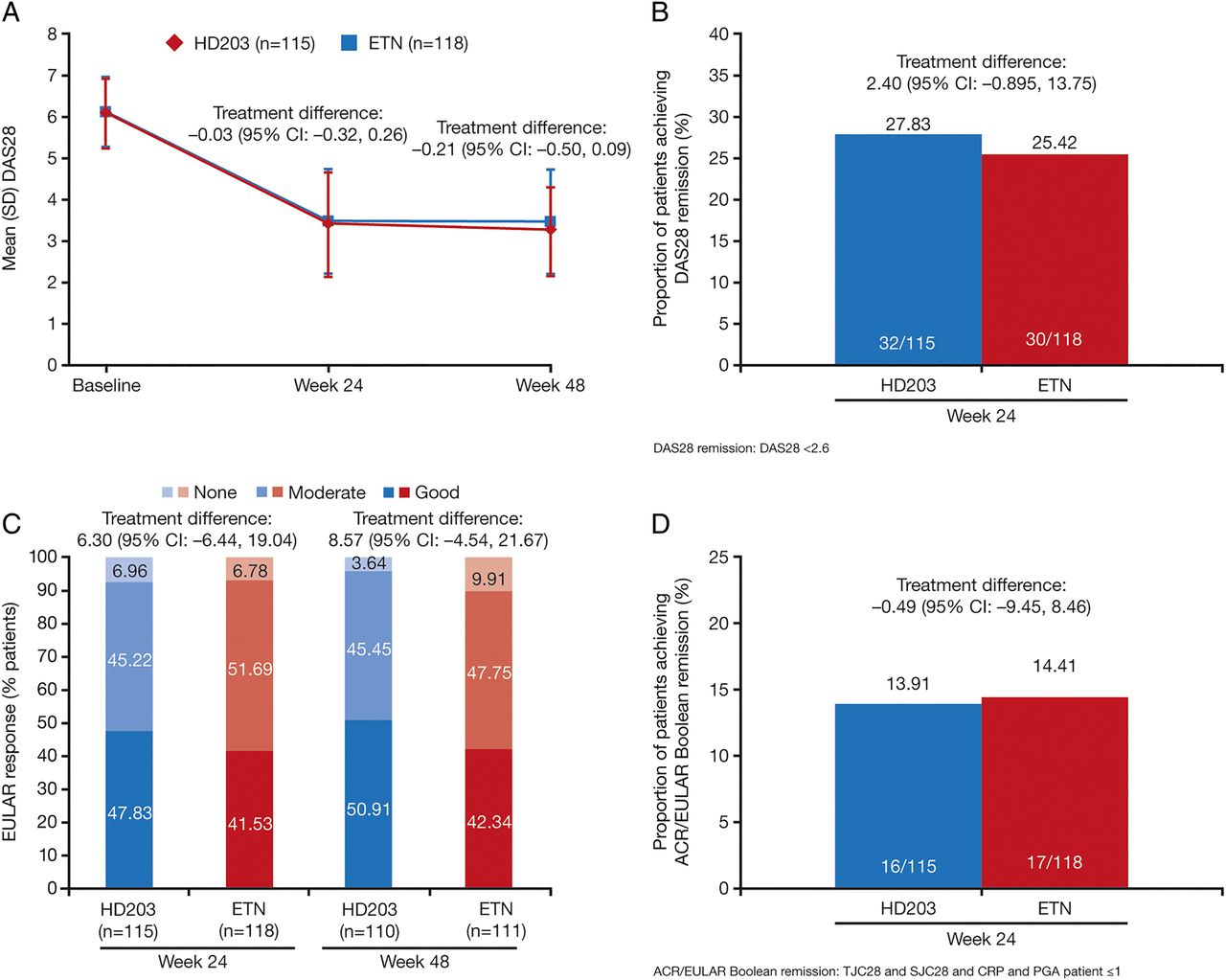
The magnitude of the change from baseline in DAS28 score at week 24 or 48 did not differ significantly between the treatment groups (figure 3A and see online supplementary table S3) and there were no significant differences at any time point between the HD203 group and the ETN group for the proportions of patients achieving DAS28 remission (figure 3B and see online supplementary tables S2 and S3).

{kind=link}
{kind=link}
{kind=link}
(A) Mean (SD) DAS28 scores at baseline, week 24, and week 48 in the per-protocol set (PPS); (B) proportion of patients achieving DAS28 remission at week 24 in the PPS; (C) European League Against Rheumatism (EULAR) response rate at week 24 and week 48 in the PPS; (D) proportion of patients achieving American College of Rheumatology (ACR)/EULAR Boolean remission, week 24 in the PPS.
There was also no significant difference between HD203 and ETN for EULAR response rates at week 24 or 48. In both treatment groups, more than 90% of patients achieved a moderate or good EULAR response (figure 3C and see online supplementary table S3). ACR/EULAR Boolean remission, either in the PPS population (figure 3D and see online supplementary table S2) or FAS (see online supplementary table S3), was also not significantly different.
The magnitude of the least squares mean change from baseline in CDAI did not differ between the HD203 and ETN groups at week 24 (–22.11 vs –22.12, respectively, treatment difference 0.01 (95% CI −1.73 to 1.76)), although it reached statistical significance at week 48 (–24.22 vs –22.44, respectively, treatment difference −1.78 (95% CI −3.53 to −0.03)) in the PPS (see online supplementary table S2). Similarly, there was no significant difference between the HD203 and ETN groups for least squares mean change from baseline in SDAI at week 24 (−23.58 vs −23.38, respectively, treatment difference (95% CI) −0.20 (−2.02 to 1.61)), while statistical significance was just reached at week 48 (–25.70 vs –23.82, respectively, treatment difference −1.88 (−3.63 to −0.13)) in the PPS. Similar findings were noted in the FAS (see online supplementary table S3).
Safety
Overall, 76.9% of patients in the HD203 group and 78.1% of patients in the ETN group developed an AE, with less than 40% of these events (34.7% HD203, 37.0% ETN) considered drug-related (table 2).
Safety assessments
The most frequently reported AE was infection (37.4% HD203, 41.1% ETN), with no major differences in the types of AEs noted between groups (table 2), including serious AEs.
Serious AEs were experienced by 19 patients (12.9%) in the HD203 group and by 18 patients (12.3%) in the ETN group. The most common SAEs in the HD203 and ETN groups, respectively, were pneumonia (n=3; n=1), pyelonephritis (n=2; n=1) and spinal compression fracture (n=0; n=3). A total of 4/5 SAEs in the HD203 group (80%) and 3/7 in the ETN group (43%) were considered drug-related. Two deaths occurred on study, with both reported in the ETN group: one due to cerebral haemorrhage and one due to acute renal failure and sepsis. Study investigators considered that the cerebral haemorrhage event was unrelated to study treatment and the renal failure was possibly related to treatment.
Eight patients taking HD203 and three taking ETN developed ADAs over 48 weeks. Of those patients, 3/8 and 1/3 were neutralising antibody-positive. Antibody development did not impact efficacy or safety outcomes.
Patient reported outcomes
Overall changes in PROs were similar between groups (table 3 and see online supplementary table S4).
Patient-reported outcomes (PPS)
Discussion
The HERA study met the primary objective of demonstrating equivalence in efficacy of HD203 compared with ETN in patients with RA receiving MTX. This is supported by data from a range of secondary efficacy analyses. PROs, including quality of life assessments, demonstrated comparable results overall between the HD203 and ETN groups. HD203 was well tolerated, with safety comparable with that of ETN. Data presented here are supported by a phase I trial demonstrating equivalent PK profiles and comparable tolerability of HD203 compared with ETN.27
ACR20 is a well-established primary end point across pivotal RA studies and is endorsed by regulators. Equivalence margins need to be based on meta-analysis of published innovator data, as well as being clinically meaningful. Although the 20% equivalence margin was considered acceptable by Korean regulators at the time of study, more recently it has been suggested that a 15% margin may be more appropriate and is consistent with recommendations from regulatory bodies and experience from past clinical trials.32 Meta-analysis of all relevant studies published after 19995–7 ,33–37 confirms that the 40% effect size remains representative of the average effect size over time (see online supplementary appendix 2, for further details). Consequently, while the study results presented here lie well within the established 20% equivalence margin, they also hold true when the more stringent (15%) evaluation is applied. Comparison of HD203 and ETN on secondary end points, including more stringent end points such as ACR-EULAR Boolean remission rates38 and those most relevant in clinical practice also indicate that HD203 is as efficacious as ETN.
The finding that more patients in the HD203 PPS group achieved ACR50 responses at week 24 and 48 than in the ETN PPS group does not abrogate the primary finding of equivalent efficacy. This study was not powered to show significant differences for secondary end points and, because ACR20 and ACR50 response groups are not mutually exclusive, there is a greater chance of a type I error when analysing ACR50 and ACR70 response rates at the same time as analysing ACR20 rates.39 Post hoc analyses of mutually exclusive responder rates were undertaken on the HERA data (see online supplementary table S5) and, in these analyses, there was no significant difference in ACR response rates between the HD203 and ETN groups (p>0.05).
Between groups at baseline, fewer patients in the HD203 group (81.7%) than in the ETN group (91.5%) were positive for RF. Positivity for anti-CCP could be considered a more specific marker for the presence of RA than RF,40–44 and this was well balanced between the HD203 and ETN groups, as was the proportion of patients who were either RF or anti-CCP antibody-positive. Additional analysis was performed for RF-positive patients in the 24-week PPS assessing the proportion achieving ACR20 response, and no significant differences between groups were found (see online supplementary table S6).
HD203 and ETN showed comparable safety profiles, and the incidence and severity of AEs were not unexpected. The immunogenicity of ETN is low and, as expected, this was demonstrated for HD203 and ETN during this study. There is no evidence to indicate that the development of antibodies to ETN affects safety or efficacy.45 ,46
Conclusion
HD203 showed equivalent efficacy to ETN in patients with RA receiving MTX by meeting the primary end point of equivalent ACR20 response at 24 weeks. HD203 was well tolerated, with safety comparable with that of ETN. Few patients tested positive for ADAs in either group. In addition to the previously generated analytical, non-clinical and clinical data, including unpublished preclinical data reviewed by the Korean health authority, the HERA study confirms the biosimilarity of HD203 and reference etanercept.
Acknowledgments
The authors thank the patients and all the investigators who participated in the study. Professional medical writing and editorial assistance were provided by ACUMED, an Ashfield Company, part of UDG Healthcare.
References
Footnotes
Handling editor Tore K Kvien
Contributors All authors meet the following conditions: (1) conception and design, acquisition of data or analysis and interpretation of data, (2) drafting the article or revising it critically for important intellectual content and (3) final approval of version submitted for publication.
Funding Hanwha Chemical Biologics Co., Seoul, Republic of Korea funded the HERA study.
Competing interests S-CB has acted as a consultant for Hanwha Chemical Co. and Ares Trading SA. At the time of writing, S-RL, YA, and HYP were employees of Hanwha Chemical Biologics, Seoul, Republic of Korea. At the time of submission, S-RL was an employee of AbbVie, Seoul, Republic of Korea.
Patient consent Obtained.
Ethics approval The study was conducted in accordance with the Declaration of Helsinki and was consistent with International Conference on Harmonisation Good Clinical Practice. The protocol and patients’ informed consent received institutional review board/independent ethics committee approval prior to initiation of the study.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement The FAS dataset for American College of Rheumatology response and ACRn is available on request for reviewer use only.
Linked Articles
- Editorial
- Clinical and epidemiological research
- Clinical and epidemiological research