Article Text

Abstract
Objectives To compare the efficacy, safety, immunogenicity and pharmacokinetics (PK) of SB2 to the infliximab reference product (INF) in patients with moderate to severe rheumatoid arthritis (RA) despite methotrexate therapy.
Methods This is a phase III, randomised, double-blind, multinational, multicentre parallel group study. Patients with moderate to severe RA despite methotrexate therapy were randomised in a 1:1 ratio to receive either SB2 or INF of 3 mg/kg. The primary end point was the American College of Rheumatology 20% (ACR20) response at week 30. Inclusion of the 95% CI of the ACR20 response difference within a ±15% margin was required for equivalence.
Results 584 subjects were randomised into SB2 (N=291; 290 analysed) or INF (N=293). The ACR20 response at week 30 in the per-protocol set was 64.1% in SB2 versus 66.0% in INF. The adjusted rate difference was −1.88% (95% CI −10.26% to 6.51%), which was within the predefined equivalence margin. Other efficacy outcomes such as ACR50/70, disease activity score measured by 28 joints and European League against Rheumatism response were similar between SB2 and INF. The incidence of treatment-emergent adverse events was comparable (57.6% in SB2 vs 58.0% in INF) as well as the incidence of antidrug antibodies (ADA) to infliximab up to week 30 (55.1% in SB2 vs 49.7% in INF). The PK profile was similar between SB2 and INF. Efficacy, safety and PK by ADA subgroup were comparable between SB2 and INF.
Conclusions SB2 was equivalent to INF in terms of ACR20 response at week 30. SB2 was well tolerated with a comparable safety profile, immunogenicity and PK to INF.
Trial registration number NCT01936181.
- Rheumatoid Arthritis
- Anti-TNF
- DMARDs (biologic)
- Disease Activity
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune inflammatory disease that leads to morbidity resulting in high societal costs.1 ,2 While disease modifying antirheumatic drugs such as methotrexate (MTX) have significantly improved the outcome in RA, not all patients respond.3 The advent of biological agents including tumour necrosis factor (TNF) inhibitors has revolutionised the treatment of RA;3 ,4 however the high cost is a significant burden to the patient and society.5
A biosimilar is a biologic agent that contains a (similar) version of the active substance of an already authorised original biological medicinal (reference) product.6 Due to the complexity of the manufacturing process, biosimilars differ from generic drugs in the chemical drug area.6 ,7 Thus, the approval pathway of biosimilars is different from generics; very roughly three major steps are employed.8 First, a comprehensive physicochemical and biological characterisation6 is done to prove similarity on the molecular level (including in vivo and in vitro assays), second, a pharmacokinetic (PK) study is done to show bioequivalence, and finally, an efficacy study (usually a randomised controlled study) is done to demonstrate clinical equivalence, compared with the reference product. The development of Remsima (code name CT-P13, Celltrion, Incheon, Korea), a biosimilar of infliximab (Remicade, Janssen Biotech, Horsham, Pennsylvania, USA), has followed this process9–11 and recently been approved by the European Medicines Agency.12 The development of biosimilars is anticipated to greatly decrease the economic burden of biological therapy.13
SB2 is developed as a biosimilar of infliximab. SB2 has undergone the stepwise process described above; SB2 was shown to be similar on the molecular level and bioequivalent in normal human subjects in a phase I PK study,14 all compared with the infliximab reference product (INF). This study now reports the primary results of the phase III study—to demonstrate clinical equivalence in patients with moderate to severe RA despite MTX treatment, compared with INF.
Patients and methods
Patients
Patients who were 18–75 years old with RA classified by the 1987 American College of Rheumatology (ACR) classification criteria for RA were enrolled; patients had to have had RA for at least 6 months with least six tender joints and six swollen joints; an erythrocyte sedimentation rate (ESR) of ≥28 mm/h or a C reactive protein of ≥1.0 mg/dL was required. Patients had to take MTX for at least 6 months and had to be under a stable dose for at least 4 weeks before randomisation. For details of inclusion and exclusion criteria, see online supplementary appendix S1.
Study design
This study is a phase III, randomised, double-blind, multinational, multicentre parallel group study (NCT01936181, EudraCT 2012-005733-37). The study consists of a 54-week main study and an additional 24-week transition (switching) study; this report is about the results of the 54-week main study up to week 30 (for the graphical presentation see online supplementary appendix S2-1), which includes the primary outcome. Patients were randomised in a 1:1 ratio to receive either SB2 or INF of 3 mg/kg intravenously. Randomisation and treatment allocation was implemented through an interactive web responsive system (Cenduit LLC, see online supplementary appendix S3-1). Infusion of SB2 or INF was done over 2 h; dosing was done at each visit at week 0, week 2, week 6, week 14, week 22, week 30, week 38 and week 46. Dose increases could occur from week 30 by 1.5 mg/kg per visit, up to a total of 7.5 mg/kg. The final visit for the main study occurred at week 54. To prevent infusion related reactions (IRRs), premedications such as corticosteroids, antihistamines or paracetamol were allowed per investigator discretion. MTX was given as an oral or parenteral weekly dose of 10–25 mg/week with folic acid of 5–10 mg/week. Non-steroidal anti-inflammatory drugs and corticosteroids (≤10 mg prednisolone) were allowed if taken for a stable dose for 4 weeks before randomisation. Other disease modifying antirheumatic drugs except for MTX were prohibited during the study. All patients were screened for tuberculosis (TB) by medical history, chest X-ray and QuantiFERON-TB Gold In-Tube tests (Qiagen); QuantiFERON tests were done at screening, week 22 and week 54. Patients with active TB were ineligible for the study and those who were found to have latent TB had to undergo prophylaxis according to country-specific guidelines to enrol in or continue with the study. The study was conducted in 73 centres in 11 countries from Europe and Asia. The study was conducted according to the Declaration of Helsinki and Good Clinical Practice issued by the International Committee on Harmonisation. All patients gave formal written informed consent before participating in the study.
Assessments
Efficacy, safety and immunogenicity assessments for all patients were done at each visit before SB2 or INF infusion.
The primary end point of the study was the ACR 20% (ACR20) response at week 30 in the per-protocol set (PPS). Other secondary efficacy end points included ACR50 and ACR70, disease activity score measured by 28 joints-erythrocyte sedimentation rate (DAS28-ESR) and European League against Rheumatism (EULAR) response. A post hoc analysis of simplified disease activity index (SDAI) and clinical disease activity index (CDAI) was done to measure the proportion of patients achieving low disease activity (LDA) or remission.15 ,16 Efficacy components such as tender and swollen joint counts, visual analogue scales scores and health assessment questionnaire of disability index scores were assessed before each infusion.
Safety assessments included monitoring of vital signs, physical examination, laboratory assessments and reports of adverse events (AEs). AEs were collected in particular for serious AEs, serious infections or TB and IRRs.
Immunogenicity assessments were done by measuring serum antidrug antibodies (ADAs) to infliximab at each visit before infusion. ADA-positivity was defined as those who had at least one positive ADA result up to week 30. This was a prespecified outcome, according to recommendations from the American Association of Pharmaceutical Scientists.17 It accommodates all measures of ADA incidence over each individual time point that may be subject to variation. Those who were ADA-positive were additionally assessed for neutralising antibodies. A single-assay approach with a SB2 tag was used to assess immunogenicity. ADAs were measured using validated electrochemiluminescence immunoassays and neutralising antibodies were measured using a competitive ligand-binding assay (MesoScale Discovery platform, Meso Scale Discovery, Rockville, Maryland, USA).
PK assessments were done by measuring the serum trough concentrations (Ctrough) of infliximab before each infusion. Serum infliximab concentrations were determined using a validated ELISA.
Sample size and statistical analysis
The primary objective was to demonstrate equivalence of ACR20 at week 30. To determine equivalence between SB2 and INF the 95% CI of the ACR20 rate difference had to be within the prespecified margin of −15% and +15%. The equivalence margin was determined using data from several INF studies4 ,18 ,19 and regulatory guidelines.20 ,21 Sample size was calculated assuming this equivalence margin of ±15%, an effect size of 57% and a 20% dropout rate. With a significance level of 5% and a power of 80%, a sample size of at least 292 randomised patients per treatment group was required in order to reach the required subject size for the PPS.
The primary efficacy outcome was analysed using the PPS and the full analysis set (FAS).22 FAS follows the same principles of the intention-to-treat analysis; FAS included all randomised patients who received at least one dose of SB2 or INF. In addition, if missing data occurred, such patients were assumed to be ACR20 non-responders in FAS. For analysis of ACR20, the rate difference was adjusted by baseline C reactive protein and geographical region using a non-parametrical analysis of covariance.23–25 Analysis of ACR50 and ACR70 was also done in PPS and FAS; DAS28, SDAI, CDAI and EULAR response were done only in the FAS (only available DAS28 and SDAI/CDAI were analysed in this case). Subgroup analysis of ACR20 was done by comparing ACR20 response rates within each ADA subgroup (positive or negative) in a prespecified manner. To formally test the differential influence of ADA on SB2 or INF, an analysis of covariance was done including an ADA-by-treatment interaction term in the model.
A prespecified exponential time-response model using non-linear mixed models26 was fitted to compare the ACR20 response between SB2 and INF over time (see also online supplementary appendix S3-2). The squared differences across all time points from the two curves (2-norm) were measured, and if the upper limit of the 95% CI of the 2-norm was less than 61.80, the two curves were considered equivalent.
Safety results were presented as the number of patients with percentage who had a particular AE in the safety analysis set (SAF; those who received at least one dose of SB2 or INF). Immunogenicity results were presented as the number of patients with percentages having incident ADA up to week 30 from the SAF. PK assessment was done in the PK population (approximately the first enrolled 50% of the study population) up to week 30. PK results are shown as mean Ctrough with SD and coefficient of variation from the PK population.
General statistical analysis was done using SAS V.9.2 (SAS, Cary, North Carolina, USA). PK parameters were calculated by non-compartmental analyses (WinNonlin V.5.2, Pharsight, Mountain View, California, USA). Graphical figures were made using R 3.0.1 (http://www.r-project.org).
Results
Patients
The study was conducted from August 2013, and the results presented in this report are from data that were collected up to mid-November 2014. The median follow-up period was 296 days. This data set included all patients that completed the week 30 visit. Among 805 patients screened, 584 patients were randomised (SB2, N=291 and INF, N=293, figure 1). From the SB2 treatment group, one patient was found to be ineligible after randomisation and withdrew before the first infusion. The baseline characteristics of the study population are shown in table 1; the two treatment groups were well-balanced. Two hundred and forty-six patients from the SB2 treatment group and 259 patients from the INF treatment group completed the week 30 visit; the most common reason for withdrawal was due to AEs and withdrawal of consent (figure 1). Among those who completed week 30, 15 patients (5.2%) from SB2 and 12 patients (4.1%) from INF were excluded from the PPS due to protocol deviations (see online supplementary appendix S4–3).
Baseline characteristics of the study population

Disposition flow chart of the study population. Among the 584 randomised, one patient from the SB2 treatment group withdrew before taking the first dose of SB2. Accordingly, the full analysis set (FAS) for SB2 is N=290 and infliximab reference product (INF) N=293 (same with the safety population (SAF)). The per-protocol set (PPS) for SB2 is N=231 and INF N=247. The pharmacokinetics population for SB2 is N=165 and INF N=160 (approximately 50% of the FAS).
Efficacy
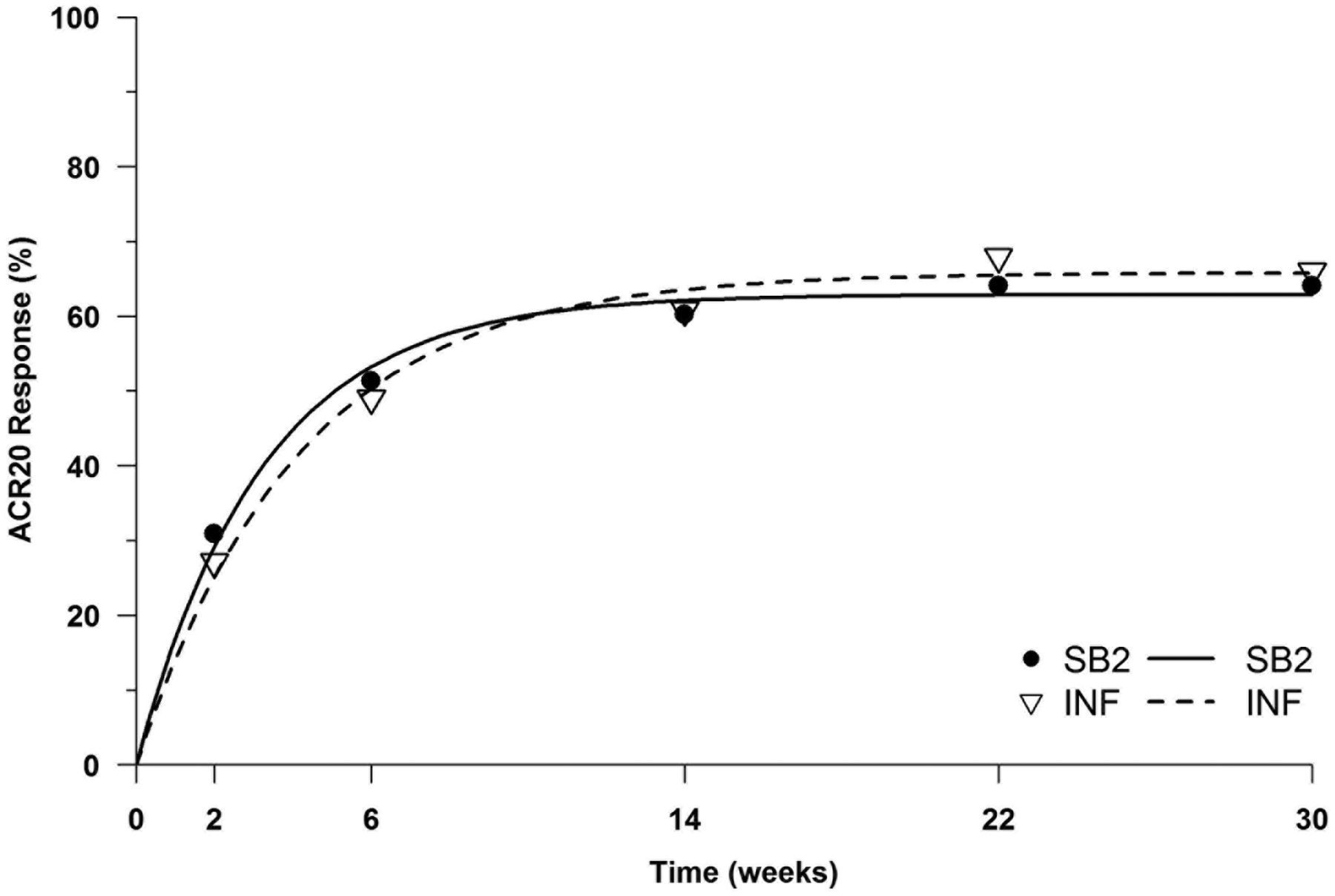
The primary efficacy end point ACR20 at week 30 is shown in figure 2. The ACR20 for the PPS was 64.1% for SB2 and 66.0% for INF. The 95% CI for the rate difference was −10.26% to 6.51%, which was within the prespecified equivalence margin of ±15%. This was also similarly shown in the FAS; ACR20 was 55.5% for SB2 and 59.0% for INF, with the 95% CI −10.88% to 4.97%. Thus, the equivalence of SB2 compared with INF was concluded (for unadjusted analyses, see online supplementary appendix S4-1). Other efficacy outcomes such as ACR50 or ACR70 were also similar in the PPS and FAS (figure 2). Finally, the ACR20 response over time is shown in figure 3. The ACR20 response at each visit was similar between SB2 and INF; the two time-response curves were determined to be equivalent.
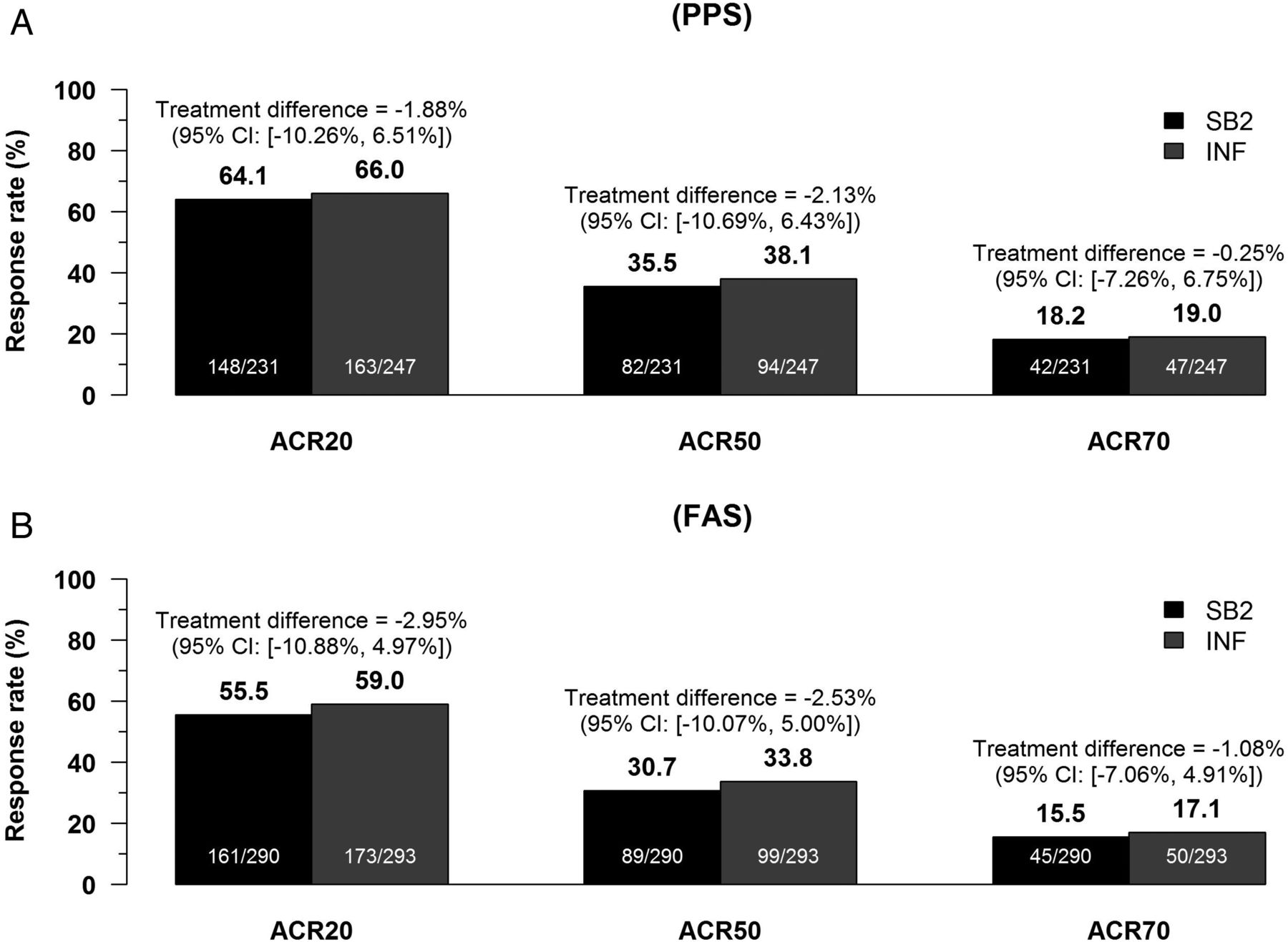
American College of Rheumatology (ACR) response rates at week 30. (A) ACR20, 50 and 70 responses for SB2 and infliximab reference product (INF) in the per-protocol set (PPS). (B) ACR20, 50 and 70 responses for SB2 and INF in the full analysis set (FAS). Rate differences were calculated by non-parametrical analysis of covariance adjusted for baseline C reactive protein and region.

ACR20 response pattern over time. Dots are actual ACR20 response rates for SB2 and infliximab reference product (INF) at each visit (per-protocol set, PPS) and the curve is fitted by non-linear mixed models employing an exponential time-response model. The upper limit of the 95% CI for the 2-norm was 35.8, which was below the prespecified equivalence margin of 61.8. For details about determining equivalence between the two time-response curves, please refer to the text.
The changes of each efficacy component used for calculating ACR responses or DAS28 activity from baseline to week 30 were similar between SB2 and INF (see online supplementary appendix S4-2). The overall ACR20 response rate was lower in the ADA-positive subgroup compared with the ADA-negative subgroup, but was also similar between SB2 and INF within each ADA subgroup (73.1% vs 73.6%, ADA-negative subgroup; 56.7% vs 58.7%, ADA-positive subgroup, see online supplementary appendix S4-3), and the interaction of ADA status by treatment group was not significant (p=0.989).
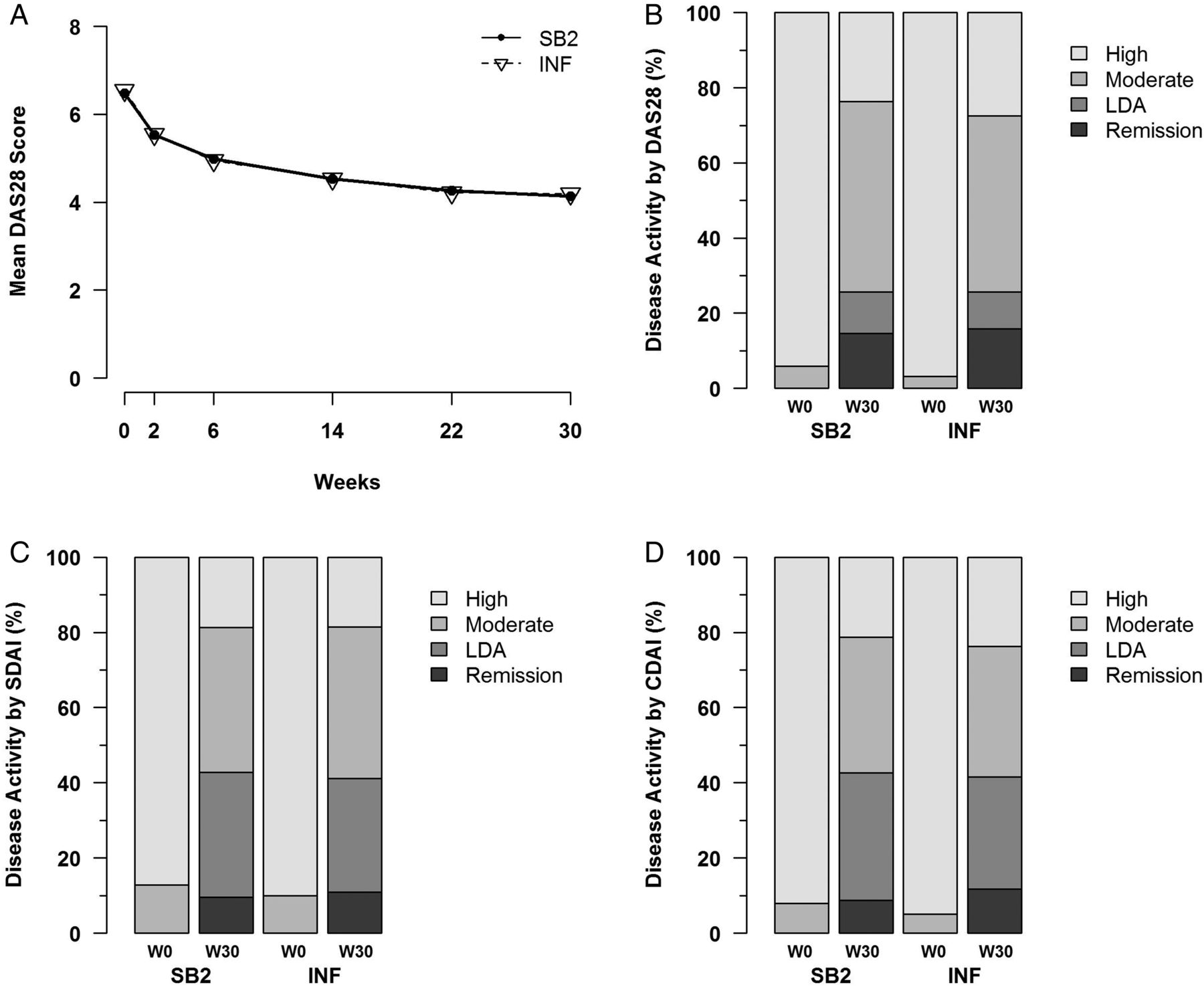
The response for DAS28 over time, the proportion of LDA/remission by DAS28, SDAI and CDAI are shown in figure 4. The improvement of DAS28 over time at each visit was similar between SB2 and INF (figure 4A), and the proportion of LDA was 11.1% for SB2 and 9.8% for INF (DAS28) and 33.3% vs 30.2% (SDAI), and for remission it was 14.6% vs 15.9% for DAS28 and 9.5% vs 10.9% for SDAI (figure 4B, C). The proportion of EULAR response was also similar between SB2 and INF (see online supplementary appendix S4-4). Overall, the efficacy end points were equivalent or similar between SB2 and INF.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
DAS28, SDAI and CDAI responses. (A) Mean DAS28 scores by visit for SB2 and infliximab reference product (INF). (B–D) Disease activity classification by DAS28, SDAI and CDAI. Remission is defined as DAS28<2.6, SDAI≤3.3 or CDAI≤2.8 and LDA is defined as DAS28 2.6≤to<3.2, SDAI 3.3<to≤11.0 or CDAI 2.8<to≤10.0. DAS28, disease activity score measured by 28 joints; SDAI, simplified disease activity index; CDAI, clinical disease activity index; LDA, low disease activity.
Safety
During the study period 499 treatment-emergent AEs (TEAEs) occurred in 167 patients (57.6%) in the SB2 treatment group and 529 TEAEs occurred in 170 patients (58.0%) in the INF treatment group (table 2). The most common TEAEs that occurred were latent TB, increased alanine aminotransferase (ALT) levels and headache. Most of the TEAEs were mild to moderate in severity. The proportion of TEAEs reported as related to the study drug was low (SB2 21.4% vs INF 20.1%). Most of the patients with treatment-emergent latent TB underwent TB prophylaxis and none of them developed TB.
Display of adverse events
Among the TEAEs, 9.0% from SB2 and 8.9% from INF reported at least one serious AE. There were nine patients (3.1%) from SB2 who developed a serious infection or TB compared with six (2.0%) patients in the INF treatment group (incidence rate: 4.1 cases/100 person-years for SB2 and 2.7 cases/100 person-years for INF), and one event in each treatment group was active TB (table 2). None of the active TB cases were found to have latent TB at screening. No serious cases of opportunistic infections were reported. There were 28 patients (4.8%) who developed an IRR; 15 (5.2%) from SB2 and 13 (4.4%) from INF reported an IRR. The number of patients who developed IRRs by ADA status were 13 (4.5%) for SB2 and 9 (3.1%) for INF in the ADA-positive subgroup and 2 (0.7%) and 4 (1.4%) in the ADA-negative subgroup. There were no reported cases of delayed hypersensitivity or serum sickness. There were two cases of malignancies from SB2 (prostate cancer and breast cancer) and one case of heart failure from INF, which was also the only case of death. Overall, the safety profile was comparable between SB2 and INF.
Immunogenicity and PKs
Patients who developed ADA up to week 30 were 55.1% (158/287) in the SB2 treatment group and 49.7% (145/292) in the INF treatment group, which difference was not statistically significant (p=0.212).
The PK results from the PK population are shown in online supplementary appendix S5. Overall, the Ctrough of infliximab was similar between SB2 and INF over time and was also similar within each ADA subgroup (ADA-positive and ADA-negative) between SB2 and INF (data not shown).
Discussion
The results from this randomised, double-blind study demonstrate the equivalence of efficacy between SB2 and INF as well as the comparability in safety, immunogenicity and PK profiles. The results are comparable to the previous PLANETRA study,11 which also studied a biosimilar of infliximab in a similar setting. The primary end point ACR20 has been compared in various ways to demonstrate the robustness of equivalence; the results uniformly exhibit the equivalence of ACR20 between SB2 and INF. In particular, our study has compared the efficacy end points at all visits, and has demonstrated the equivalence of ACR20 response over time, which is an advance over the data presented for the PLANETRA study. The comparability of efficacy end points over time has been suggested as an important criterion for biosimilarity,27 and our results are supportive in such manner. It is also notable to observe that other efficacy outcomes besides the ACR response such as DAS28, SDAI, CDAI and EULAR responses all show similarity between SB2 and INF, further supporting the biosimilarity of SB2 to INF. While results concerning SDAI and CDAI were post hoc analyses, as the components of these indices are all included within the components of ACR20 or DAS28, any bias due to post hoc specifications are expected to be minimal.
In terms of safety, SB2 has demonstrated a comparable safety profile to that of INF; in particular, the incidence of AEs associated with TNF inhibitors such as serious infections, TB, IRRs, malignancies and heart failure were comparable between SB2 and INF. The incidence rate of serious infections or TB was comparable to previous studies.28 ,29 It is notable that the incidence of TB was low (one case for each treatment group), which universal TB prophylaxis for patients with latent TB at screening might have contributed to.
The incidence of increased ALT levels was higher in SB2 than in INF (7.9% vs 2.7%, table 2), however, patients whose laboratory ALT values with an ALT of 3×ULN (upper limit of normal) and 5×ULN were 5.2% and 1.4%, respectively, for SB2, which are within the historically reported range of ALT abnormalities with INF (2.5–9.5% and 0.6–3.6%, respectively).30
Immunogenicity was comparable between SB2 and INF; the incidence of ADA (∼50%) is higher than previous INF pivotal trials4 but comparable with recent INF studies31 ,32 and PLANETRA (∼48%), probably reflecting the advance of assay technology during the time.11 The apparent numerical difference was not statistically significant and efficacy was similar within each ADA subgroup (see online supplementary appendix S4-2), a pattern that was also observed in IRRs. The finding that efficacy is lower and the risk of IRRs is higher in ADA-positive patients is consistent with prior experience with infliximab.11 ,31 ,32 In ADA-positive patients we observed an approximately 40% higher rate of infusion reactions for SB2 compared with INF; however, among the ADA-negative patients, infusion reactions were twice as high for INF than SB2. The overall rate of infusion reactions was similar with 5.2% for SB2 and 4.4% for INF. Long-term observation will allow further insights into this important aspect. The PK results showed a comparable distribution of mean Ctrough and variance to previous infliximab studies.4 ,11
As mentioned, biosimilars are hoped to decrease the economic burden in the treatment of RA. The issue of cost-effectiveness of biologics5 may have to be addressed again with the advent of biosimilars,33 which could also have significant influence on local reimbursement policies.
While long-term efficacy and safety profiles and pharmacovigilance in the postmarket setting are important considerations7 and will have to be obtained, these are not within the scope of this report. However, to address these aspects at least in part, the main study is continuing with a 54-week end point for assessing long-term efficacy and safety including radiographic damage.
In conclusion, from the results of this randomised, double-blind study, SB2 has demonstrated clinical equivalence to INF in terms of ACR20 at week 30; other efficacy end points also show consistently similar findings when compared with the originator product. The safety, immunogenicity and PK profiles are comparable between SB2 and INF.
Acknowledgments
The authors thank the patients who were involved in this study, the study personnel who made this work possible, and the study investigators: Bosnia and Herzegovina: Sokolovic S, Mekic M, Prodanovic N; Bulgaria: Dimitrov E, Geneva-Popova M, Mihaylova M, Staykov I, Toncheva A, Penev D, Oparanov B; Czech Republic: Dokoupilova E, Galatikova D, Ciferska H, Vitek P, Janska L; Korea, Republic of: Shim SC, Kang YM, Kim HA, Choe J-Y, Lee S-H, Bae S-C, Kim J, Kwok S-K, Lee YJ, Lee S-K; Latvia: Kadisa A, Mihailova A, Saulite-Kandevica D, Saleniece S; Lithuania: Milasiene R, Baranauskaite A, Arstikyte I, Basijokiene V; Philippines: Santos Estrella P, Hao L, Manapat-Reyes BH, Eullaran R; Poland: Porawska W, Kolczewska A, Stasiuk B, Janecka I, Grabowicz-Wasko B, Jedrychowicz-Rosiak K, Leszczynski P, Ruzga Z, Rychlewska-Hanczewska A, Hajduk-Kubacka S, Hilt J, Niebrzydowski J, Zielinska A; Romania: Berghea F, Popoviciu H, Mirea G, Pavel M, Ieremia G, Tanasescu C; Ukraine: Rekalov D, Zhdan V, Povoroznyuk V, Ignatenko G, Ter-Vartanian S, Vatutin M, Stanislavchuk M, Gnylorybov A, Golovchenko O, Yatsyshyn R, Tseluyko V, Yagensky A, Iaremenko O, Shevchuk S; UK: Ong V, Mckay N, and the study team: Ilsun Hong (Samsung Bioepis).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
A portion of the manuscript was presented as an abstract (SAT0152) at Rome in EULAR 2015.
Correction notice This article has been corrected since it was published Online First. Minor corrections have been made to the author affiliations, funding section and table 2.
Contributors JSS, JC and YHR contributed to study conception and design, acquisition of data, analysis and interpretation of data, drafting the manuscript and revising it critically for important intellectual content, and final approval of the version to be published. J-YC contributed to acquisition of data, analysis and interpretation of data, drafting the manuscript and revising it critically for important intellectual content, and final approval of the version to be published. NP, JN, IS, ED, AB, RY, MM, WP, HC, KJ-R and AZ contributed to acquisition of data, drafting of the manuscript and revising it critically for important intellectual content, and final approval of the version to be published. YHR wrote the paper on behalf of the sponsor. There were no professional writers or companies hired for this purpose. All coauthors contributed significantly to the writing of the manuscript.
Funding This study was funded by Samsung Bioepis Co., Ltd.
Competing interests J-YC reports receiving grant/research support and consultant fees from Samsung Bioepis. JSS reports receiving grant/research support from AbbVie, Jassen, MSD, Pfizer, Roche, UCB, Consultant for: AbbVie, Amgen, AstraZeneca, Astro-Pharma, Celgene, GSK, Jassen, Lilly, Medimmune, MSD, Norvartis-Sandoz, Novo Nordisk, Pfizer, Roche, Samsung Bioepis, Sanofi, UCB. AB reports receiving grant/research support from AbbVie and Samsung Bioepis. NP, JN, IS, ED, RY, MM, WP, HC, KJ-R and AZ report receiving grant/research support from Samsung Bioepis. JC and YHR are employees of Samsung Bioepis.
Ethics approval Each national regulatory agency and central or local ethical committee.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial
- Clinical and epidemiological research
- Clinical and epidemiological research