Article Text

Abstract
Objectives Current evidence suggests that immune events in the gut may impact joint inflammation in ankylosing spondylitis (AS) but the expression of gut-related trafficking molecules in the inflammed joint is poorly characterised. We aimed to (1) assess differential expression patterns of trafficking molecules between patients and controls, (2) generate joint-specific cellular signatures and (3) obtain transcriptomic profiles of noteworthy cell subpopulations.
Methods Male subjects under 40 years of age fulfilling the mNY criteria were recruited. The following cells were surface stained using a 36-marker mass cytometry antibody panel: (1) peripheral blood mononuclear cells from AS patients, and healthy controls; (2) synovial fluid mononuclear cells from AS and rheumatoid arthritis (RA) patients. Additionally, RNA-seq was performed on CD8+ T cell subpopulations from the synovial fluid (SF).
Results Mature CD8+ T cells were enriched in AS SF, with a distinct pattern of integrin expression (β7, CD103, CD29 and CD49a). RNA-seq analysis of SF-derived CD103+CD49a+CD8+ T cells revealed elevated TNFAIP3, GZMB, PRF1 and IL-10.
Conclusions We have identified a novel integrin-expressing mature CD8+ T cell population (CD49a+CD103+β7+CD29+) that appears to be more prevalent in AS SF than RA SF. These cells seem to possess dual cytotoxic and regulatory profiles which may play a role in AS pathogenesis.
- Ankylosing Spondylitis
- CyTOF
- RNA-seq
- integrins
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Axial spondyloarthritis (AS) has clinical, genetic and immunological overlaps with inflammatory bowel disease, thereby making the gut–joint axis of inflammation an emerging area of research and therapy.
Phenotypic knowledge of immune cells, implicated in inflammation of the gut and joint, is unclear.
What does this study add?
Proteomic and transcriptomic analyses helped identify a mature CD8+ T cell subpopulation, enriched in gut-associated trafficking molecules (CD49a+CD103+β7+CD29+), which was elevated in AS synovial fluid.
This supopulation appeared to have a dual cytotoxic and regulatory profile, which may play a role in AS pathogenesis.
How might this impact on clinical practice or future developments?
Therapeutic intervention using integrin-based blocking agents have had inconsistent results in proof-of-concept studies in AS patients, mandating a more thorough understanding of trafficking molecules in disease pathogenesis.
Introduction
Ankylosing spondylitis (AS), is characterised by chronic inflammation of the axial skeleton and often coexists with inflammatory bowel disease (IBD).1 AS prevalence ranges from 0.1% to 1.4%1 and male sex influences clinical disease expression.2 HLA-B27, an major histocompatibility complex (MHC) class I molecule, remains the strongest genetic risk factor for AS, with 80%–90% AS patients testing positive for HLA-B27.1 Since there is a strong clinical and genetic association between AS and IBD, the gut–joint axis of inflammation has become an important area of research.3 One hypothesis states that inflammation iniating in the gut is transferred to the joint due to the abberant trafficking of immune cells. Nonetheless, the trafficking molecules shared between cells in the gut and joint are poorly defined.
Leukocytes employ various trafficking molecules, such as integrins and chemokine receptors, to orchestrate their exit from the circulation and retention in tissue.4 Adhesion molecules and chemokine receptors expressed by immune cells programme targeting to specific tissues. To date, studies have identified shared trafficking molecules between gut and joint tissue. For example, α4β7, the prototypic gut homing integrin, is also expressed by immune cells in synovial tissues.5 The α4β7 ligand, MAdCAM-1, is expressed on endothelial cells at sites such as joints, eyes, skin and liver.6 Moreover, mucosal lymphocytes isolated from IBD patients bind to inflamed synovial vessels in vitro via distinct adhesion receptors.4 7
Integrin-blocking agents, approved for IBD, have been used in AS patients with coexisting IBD with conflicting case reports with regards to affects on arthritis.8–12 A major limitation to our understanding as to whether integrin blockade could be effective or exacerbative of arthritis is our incomplete understanding of the trafficking molecules associated with AS. For this reason, we screened gut-related trafficking molecule expression at the cellular level in AS patients using multidimensional mass cytometry (CyTOF). We sought to identify trafficking marker combinations, using multiple computational methods, to identify novel cell populations in AS. We demonstrate disease specificity of trafficking marker expression to the joint and we have identified a population of mature CD8+ T cells that expresses CD103 (αE integrin), β7, CD29 (β1 integrin) and CD49a (α1 integrin) and which are enriched in AS. These integrin-expressing (InEx) cells possess a dual cytotoxic and regulatory transcriptomic profile, as evidenced by elevated TNFAIP3, GZMB, PRF1 and IL-10.
Methods
Patient cohorts. CyTOF, RNA sequencing, NanoString, protein–protein interactions and cytokine multiplex assay
Detailed experimental procedures are provided in the online supplementary files 1; 2.
Results
Broad cell population changes in SFMC compared with PBMC
We began analysis of our CyTOF samples using spanning-tree progression analysis of density-normalized events (SPADE) on all live cells (selections strategy displayed in online supplementary figure S1). SPADE is an unsupervised hierarchical clustering algorithm which provides an overview of cell populations. Major cell populations appeared similar in the peripheral blood mononuclear cell (PBMC) of patients and controls; however, differences were seen in the number of cells in each node, and in surface marker expression when comparing patient synovial fluid mononuclear cell (SFMC) to PBMC (figure 1A). These qualitative SPADE observations were used to direct FlowJo-based quantitative analysis of PBMCs and SFMCs from healthy controls (HC), AS and rheumatoid arthritis (RA) samples (figure 1B). For example, CD14+CD56- cells are expanded in synovial fluid (SF), which likely represent macrophage-like cells seen frequently on histological examination (online supplementary figure S2). CD19+ expression was similar between HC and AS blood, although the frequency of CD19+ B cells was decreased in AS-SF. Further, there was a trend towards expansion of natural killer (NK) cells in SF versus blood.
Supplemental material
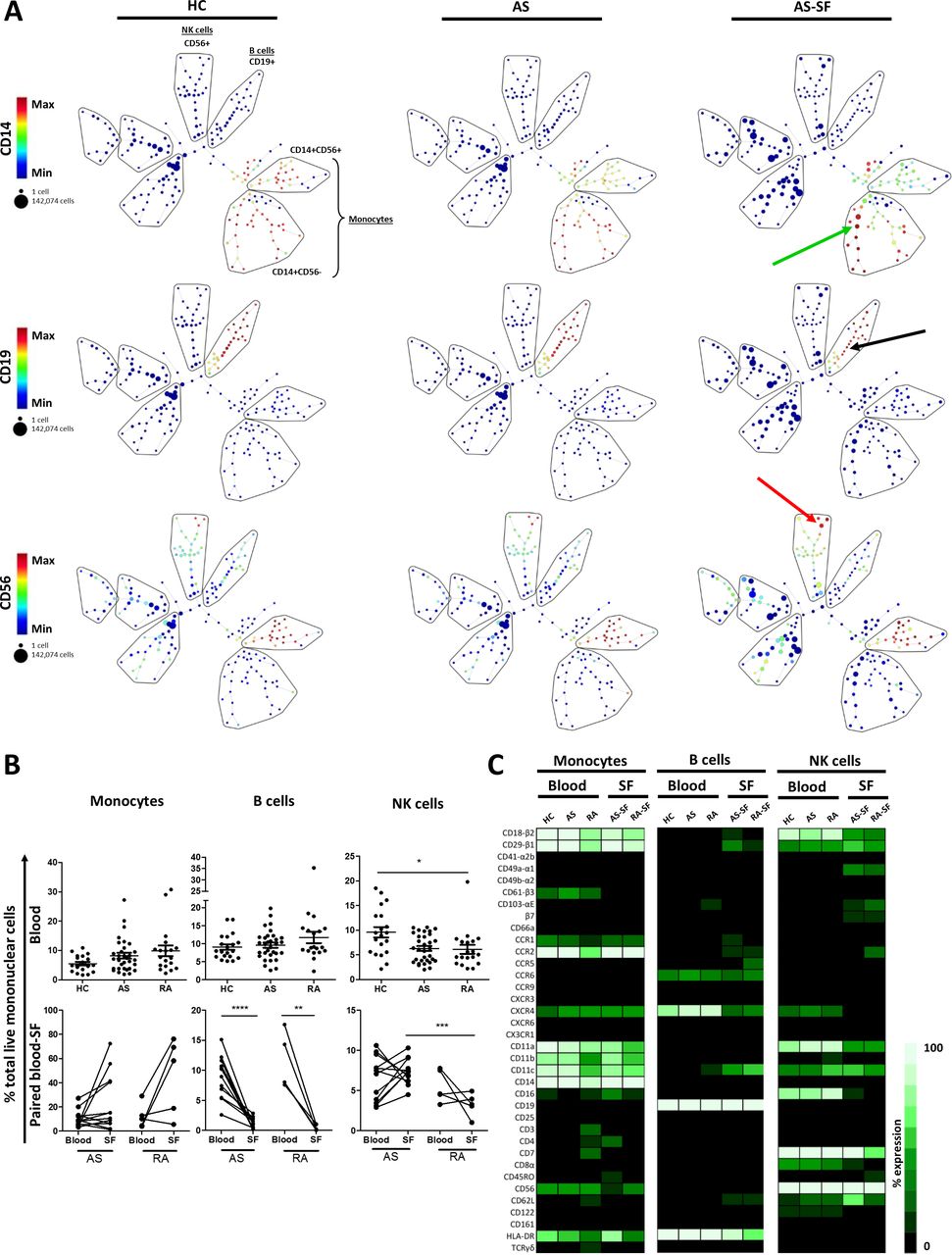
Overview of major non-T cell lineages in blood and synovial fluid using high-dimensional analyses. (A) Live single cells from HC PBMC (n=20) and AS PBMC (n=32) and AS SFMC (n=12) concatenated by group and analysed by SPADE. Pregating strategy given in online supplementary figure S1. Common lineage markers used to identify indicated cell populations (lassoed for illustrative purposes), with CD14, CD19 and CD56 expression given as examples. Arrows indicate populations highlighted in text. (B) Cell subsets were identified as follows in FlowJo: monocytes (CD14+CD19-), B cells (CD14-CD19+) and NK cells (CD14-CD19-CD3-HLADR-CD56+). HC, AS and RA blood (PBMC) data analysed by Kruskal-Wallis test with Dunns post-test. Monocytes paired blood-SF samples analysed by Wilcoxon matched pairs test, while B cells and NK cells paired blood-SF samples analysed by paired T test. AS-SF and RA-SF analysed by Mann-Whitney test. ****p<0.0001, ***p<0.001, **p<0.01, *p<0.05, n.s. p>0.05 (non-significant results not highlighted on graphs, unless indicated). (C) Monocytes, B and NK cells were gated in each sample and concatenated into a single file for each sample group using FlowJo. The expression (%+) of each marker was measured in the concatenated files, representing the mean expression per group, and were converted to a heatmap. HC PBMC (n=20), AS PBMC (n=32), RA PBMC (n=19), AS SFMC (n=12), RA SFMC (n=5). AS, ankylosing spondylitis; PBMC, peripheral blood mononuclear cell; RA, rheumatoid arthritis; SF, synovial fluid; SFMC, synovial fluid mononuclear cell.
Using heat maps, distinctive trafficking molecule expression profiles were seen on major non-T cell PBMC populations independent of disease status (figure 1C). For example, monocytes expressed CD61, CCR1 and CCR2, whereas B and NK cells did not. On the other hand, B cells expressed CCR6, while monocytes and NK cells were limited in their CCR6 expression. Notably, there were few differences in trafficking marker expression between AS and control PBMC, with respect to CD14+ monocytes, CD19+ B cells and CD56+ NK cells.
Heatmap analysis further revealed marked differences between cells in AS SF compared with blood, many of which were significant (figure 1C and online supplementary figure S3A and S3B). For example, CD14+ monocytes showed higher CD4, CD16, HLA-DR and lowered CD61 expression in SF (figure 1C and online supplementary figure S3A). CD56+ NK cells in SFMC displayed characteristically high β7, CCR1, CD49a and CD103 expression compared with PBMC, but lower CD16 and CXCR4 (figure 1C and online supplementary S3B), indicating a tissue-resident phenotype in SF.13 Some of these SF profiles were disease specific. For example, CD14+ cells in AS SF expressed more CD4+, CD16+ and HLA-DR+ than those in RA SF. Likewise, there was a trend for RA SF NK cells expressing more trafficking molecules, such as β7, CCR1 and CCR2, when compared with their AS SF counterparts.
CD56hi NK cells, a population of cytokine-expressing NK,14 were more common in the AS SF than blood and were at a higher frequency than RA SF (online supplementary figure S4A). Stratification by CD16 indicates NK cell cytotoxicity.14 CD56hiCD16+ NK cells were markedly reduced in AS SF, while CD56hiCD16- NK cells were enriched in AS SF, suggesting NK cells in SF have a less cytotoxic phenotype. The more cytotoxic14 NK cell subtype, CD56dim NK cells, were infrequent in AS versus HC blood, with a trend towards reduced frequency in AS SF. These cells appeared to be less cytotoxic, owing to the less common occurrence of CD56dimCD16+ cells and the increased CD56dimCD16+ cells (online supplementary figure S4B).
In summary, we observed few changes to non-T cell frequency and phenotype in the blood of AS patients and controls. Large changes were seen in the frequency and phenotype of these cells in the SF, highlighting the importance of studying the target site of inflammation.
Mature CD8+ T cells are enriched in AS SF
Owing to the high diversity of T cells, and their role in AS, we performed SPADE analysis by pregating on whole T cells (figure 2A). Here, it was evident that the SF was enriched with mature CD45RO+ T cells, with certain cell populations being more common in SF, as indicated by arrows in figure 2A.

Overview of major blood and synovial fluid T cell populations using high-dimensional analyses. (A) Single file representations of HC (n=20), AS PBMC (n=32) and AS SFMC (n=12) were entered into Cytobank, and gated CD3+ cells were then entered into SPADE programme. T cell subpopulations were identified on the basis of common markers, which were lassoed for illustrative purposes. Median CD45RO expression intensities per node displayed from HC and AS (PBMC) and AS-SF. Expanded populations indicated using arrows. (B) Cell subsets were identified as follows using FlowJo: T cells (CD14-CD19-CD3+HLADR-), γδ T cells (CD14-CD19-HLADR-CD3+TCRγδ+), CD4+ T cells (CD14-CD19-HLADR-CD3+CD4+) and CD8+ T cells (CD14-CD19-HLADR-CD3+CD8+). Graphs depicting T cells and γδ T cells blood samples analysed by Kruskal-Wallis test with Dunns post-test. T cells paired blood-SF samples analysed by paired T test, while γδ T cells paired samples analysed by Wilcoxon matched pairs test. CD4 and CD8 T cells blood samples analysed by one-way ANOVA with Bonferroni correction, while paired samples analysed by paired T test. AS-SF and RA-SF analysed by Mann-Whitney test. ****p<0.0001, ***p<0.001, **p<0.01, *p<0.05, n.s. p>0.05 (non-significant results not highlighted on graphs, unless indicated). (C) Mature CD4 or CD8 cells identified as CD4+CD45RO+ or CD8+CD45RO+ using FlowJo. Blood samples analysed by Kruskal-Wallis test with Dunns post-test, while paired samples analysed by paired T test. AS-SF and RA-SF analysed by Mann-Whitney test. ***p<0.001, **p<0.01, *p<0.05, n.s. p>0.05 (non-significant results not highlighted on graphs, unless indicated). PBMC, peripheral blood mononuclear cell; RA, rheumatoid arthritis; SF, synovial fluid; SFMC, synovial fluid mononuclear cell.
Statistical analysis of these major cell subsets revealed no differences in whole CD3+ T cell and γδ T cell frequencies in the blood. On the contrary, CD4+ T cells were elevated in RA patient blood, which was accompanied by a reduction in CD8+ T cells (figure 2B). RA blood CD4+ T cells had a more mature phenotype than AS and HC, and RA blood CD8+ T cells were more mature than AS (figure 2C), which may be attributable in part to the age differences in the respective patient groups.15
In SF, we observed no change in whole CD3+ T cell frequency compared with blood, however there was a reduction in CD4+ T cells and an elevation of CD8+ T cells in AS SFMC versus PBMC (figure 2B). The most striking change in the composition of T cells in blood compared with SF was the enrichment of CD45RO in both CD4+ and CD8+ T cells irrespective of disease type (figure 2C). Given this final observation, and the fact that AS is an MHCI-associated disease,1 we focused our studies on mature CD8+ T cells in the SF.
AS SF is characterised by a distinct population of CD103+CD49a+ mature CD8+ T cells
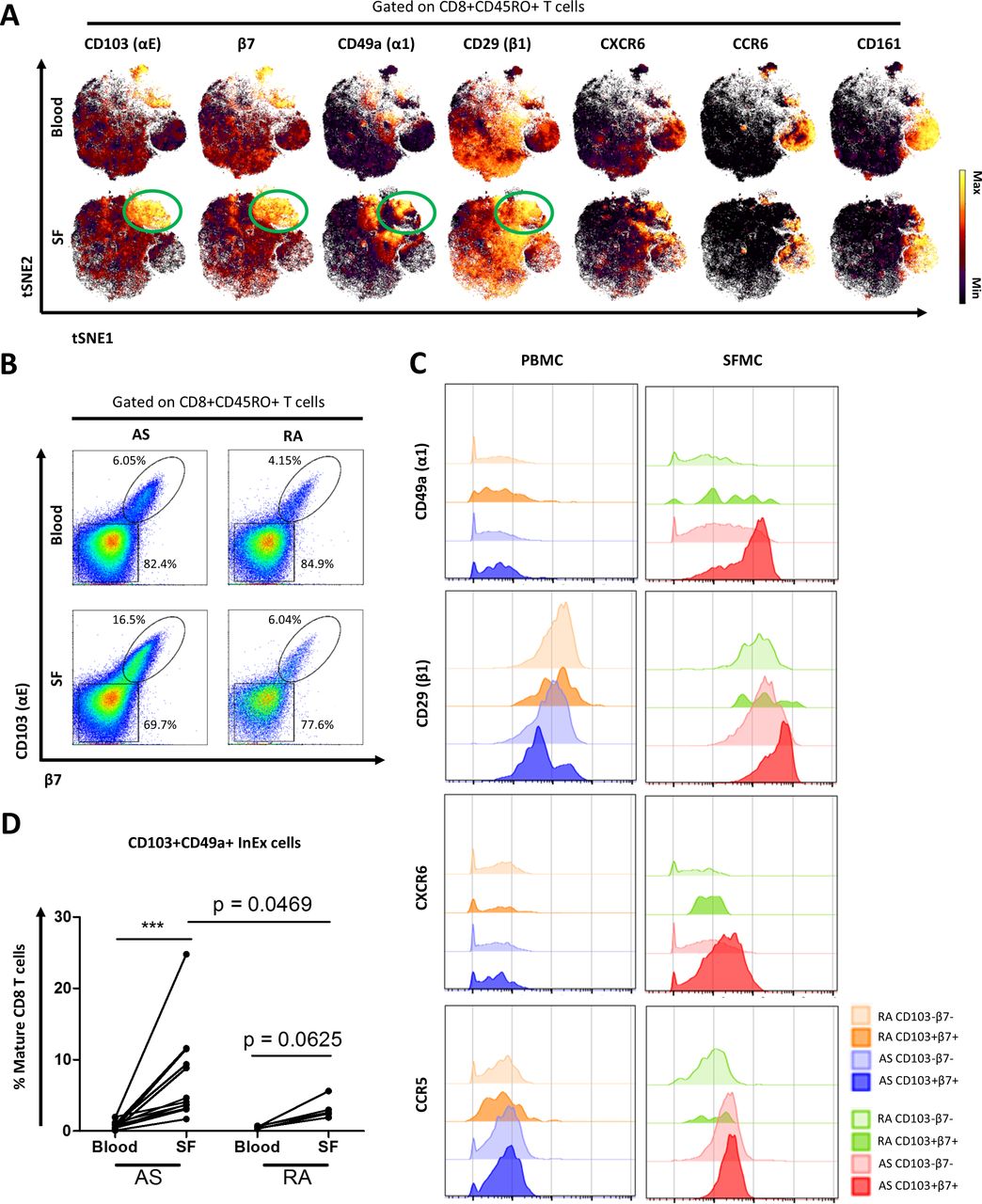
The enrichment of mature CD8+ T cells in the inflamed joint prompted a clustering analysis using the unsupervised viSNE algorithm that provides readouts at a single cell resolution. This revealed a subpopulation of mature CD8+ T cells that expressed CD103 in AS SF (figure 3A). In addition, other markers were co-expressed on CD103+CD8 T cells in SF, such as β7, CD49a, CD29 and CXCR6. These cells are likely not MAIT cells as they did not express CCR6 and CD161 (figure 3A). The enrichment of CD103+CD8+ T cells in AS SF was validated by conventional flow cytometry in an independent cohort (online supplementary figure S5A).

A population of CD103+CD49a+ (InEx) CD8 T cells is enriched in AS synovial fluid. (A) Mature CD8+ T cells from single file representations of AS PBMC (n=32) and AS SFMC (n=12) were entered into viSNE algorithm for high-dimensional analysis of trafficking molecules in as blood and SF. Trafficking molecules were selected to display their expression levels on mature CD8+ T cells. (B) Plots shown of merged single file representations of PBMC and SFMC obtained from AS and RA patients, to compare their CD103 (αE) and β7 expression patterns gated on mature CD8+ T cells. (C) Expression intensities of trafficking molecules as seen on CD103+β7+or CD103-β7- cells from mature CD8+ T cells from representative AS and RA paired PBMC-SFMC plots. (D) CD103+CD49a+ integrin expressing (‘InEx’) cells from paired blood-SF of AS and RA patients, gated using FlowJo. Paired samples analysed by Wilcoxon matched pairs test. AS SF and RA SF analysed by Mann-Whitney test. ***p<0.001. InEx, integrin-expressing; PBMC, peripheral blood mononuclear cell; RA, rheumatoid arthritis; SF, synovial fluid; SFMC, synovial fluid mononuclear cell.
Closer examination of this novel CD8+ T cells subpopulation revealed that CD103 and β7 are tightly coexpressed in both blood and SF (figure 3B). There was an enrichment of CD103+β7+ mature CD8+ T cells in AS SF versus blood (mean 17.7±2.9% vs 11.3±3.3%), and which was higher in AS SF than RA SF (online supplementary figure S5B). CD103-β7- mature CD8 T cells were reduced in AS SF compared with blood (mean 76.0±3.1% vs 84.1±3.1%) and were much lower than RA SF (online supplementary figure S5B). Further examination of these cells revealed disease-specific trafficking marker profiles: CD49a, CD29 and CXCR6 were found to be preferentially upregulated in CD103-αE+β7+ mature CD8 T cells from AS SFMC (figure 3C) whereas CXCR4 was downregulated in this cell population (online supplementary figure S5C). Trafficking markers such as CCR5, CD11a and CD18 showed no difference in expression on either CD103+β7+ or CD103-β7- mature CD8 T cells from AS or RA samples (figure 3C; online supplementary figure S5C). In addition to CD103+β7+ expression, mature CD8+ T cells from AS SF were also enriched in CD49a+CD29+ (VLA-1) expression versus blood (mean 27.5±4.4% vs 6.4±1.6%) (online supplementary figure S5D).
We designate the particular subpopulation of mature CD8+ T cells expressing CD103+β7+CD49a+CD29+ integrins as integrin-expressing (InEx) cells. Since CD103+β7+ and CD49a+CD29+ are often coexpressed we used CD103 and CD49a expression to simplify the identification of InEx cells. Indeed, using this definition, InEx cells were significantly enriched in AS SF, but barely detectable in the AS blood (mean 7.5±1.9% vs 0.9±0.2%; figure 3D). This effect was not seen between RA blood and SF paired samples. InEx cell frequency was also higher in AS SF than RA SF (mean 7.5±1.9% vs 3.1±0.6%). As a control cell population, CD103-CD49a- mature CD8+ T cells were classified as non-InEx cells and were noticeably decreased in SF versus blood of both AS and RA patients (online supplementary figure S5E). We used these definitions for subsequent studies.
As some AS and RA patients in this study were being prescribed anti-TNF biologics (Table S1), we sought to determine the influence of biological treatment on the InEx cell percentage using multiple regression analysis. In SFMC and PBMC, we discovered that biological status only predicted only 6.2% and 4.7% of the variability in the InEx cell percentages, respectively. Thus biological treatment was not a significant confounder in the immune profiling results (online supplementary figure S6A and S6B).
InEx cells possess a dual cytotoxic and regulatory transcriptome
To examine the InEx cells, their resting transcriptome was analysed using bulk RNA sequencing on fluorescence activated cell sorting (FACS)-sorted SFMC cells. Non-InEx and naïve CD8+ T cells from AS SFMC were used as comparators (online supplementary figure S7A). After filtering based on defined cut-offs (fold change greater than or less than 2 and p<0.05), there were 1124 differentially expressed genes (DEGs) (1450 including splice variants; online supplementary figure S7B) among the three groups. PCA analysis of these three populations, using the 1124 unique genes, revealed distinct transcriptomic profiles (online supplementary figure S7C). Overall, 197 genes were uniquely expressed in InEx versus non-InEx, out of which 80 were elevated and 117 downregulated (figure 4A). Specifically, InEx cells were defined by GZMB, IRF4, ITGAE, CCL5, RPTOR, TNFAIP3, BAX, PRF1 and IL-10, non-InEx cells by IRF7, CX3CR1, DUSP2, IFNAR1 and IFNAR2 and naïve CD8 T cells by GNLY, IL-17RC, S1PR1, IFNG, STAT3 and CTNNB1 (figure 4B). These observations were all significant, with considerable fold change (online supplementary figure S7D).

Transcriptomic analysis of ‘integrin-expressing’ (InEx) cell population. InEx, non-InEx and naïve CD8+ T cells were FACS purified from AS SF for RNAseq analysis (n=5 patients). (A) Resulting transcript fragments per kilobase of transcript per million (FPKM) values were filtered according to fold change greater than or less than 2 and p<0.05 cut-off and converted into Venn diagram to identify differentially expressed genes unique to or shared between each population (InEx, non-InEx and naïve). (B) Using aforementioned cut-offs, a heatmap displaying expression levels of selected genes in the three groups was plotted. Unit variance scaling was applied to rows. Rows clustered using correlation distance and average linkage. Columns clustered using Euclidean distance and average linkage. (C) 197 genes were differentially regulated in InEx versus non-InEx cells and 353 genes differentially regulated in InEx versus naïve cells. Out of these, 80 genes and 150 genes were uniquely elevated in InEx cells, respectively, and were fed into Cytoscape with ClueGo programme selected. All pathways/ontologies were selected, with network specificity ‘medium’ and p<0.05. Resulting data were converted into a graph to display pathway analysis of elevated genes. SF, synovial fluid.
We performed pathway analysis to infer function of InEx cells based on their gene expression profile. The 80 genes were involved in apoptotic signalling and in turn, lymphocyte homeostasis (BAX, ETFDH, PPP3CB, TNFAIP3), T cell receptor signalling (CD3D, LCP2, PPP3CB), ubiquitinyl hydrolase/protease activity (ALG13, OTUD1, TNFAIP3) and TNFα signalling (BAX, TNFAIP3) (figure 4C). Genes with functions such as cytokine binding (A2M, CX3CR1, FZD4, IFNAR1, IFNAR2), type I interferon activity (IFNAR1, IFNAR2) and calcium signalling pathway (CTNNB1, FZD4, ITPR2) were uniquely reduced in InEx versus non-InEx cells (online supplementary figure S7E). Pathway enrichment analysis revealed IL-10 and NFκB signalling networks as enriched in all 1124 genes, while granzyme A-mediated apoptosis pathway was enriched in InEx versus naïve (figure 4D).
To further phenotype InEx cells, we performed TCR stimulation and assessed gene expression. Compared with non-InEx cells, there was an elevation of transcripts from KLRC1, KLRC2, GNLY, CCL20, CCR6, CCRL2 and SLAMF1. Genes such as GZMK, PECAM1 and HLA-DPA1 were lower in InEx cells (figure 5).

NanoString analysis of stimulated InEx and non-InEx cell populations. FACS-sorted InEx and non-InEx cells from AS SFMCs (n=6) were stimulated with anti-CD2/CD3/CD28 T cell activation beads and 500 U/mL IL-2 for 72 hours/37°C. RNA was isolated and processed using the nCounter mRNA assay. Log2 normalised RNA transcripts were converted into fold change (FC) values (InEx/non-InEx), analysed using paired T test (p<0.05) and graphed. Dotted line indicates FC=1 (NO change). Uncorrected p values used for analysis owing to low sample numbers per group. InEx, integrin-expressing; SFMCs, synovial fluid mononuclear cells.
A protein–protein interaction network analysis was conducted in order to visualise the design of interactions between the 1124 DEGs comprehensively. Interestingly, some of the genes that define InEx cells, such as IRF4, CCL5, TNFAIP3, BAX and IL-10, encode proteins that appear to interact with each other (figure 6). Specifically, TNFAIP3 protein also seems to interact with LCP2, whose phosphorylated form acts as a substrate for ZAP-70 in early TCR-mediated signaling16 (online supplementary figure S8A). Granzyme B (GZMB) and perforin (PRF1) proteins interact with CD2, a costimulatory molecule17 (online supplementary figure S8B).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Protein–protein interaction network. The 1124 DEGs were submitted to the IID platform, from which corresponding protein-protein interactions (PPIs) and degree of interaction for each gene was obtained. These were then input into the navigator programme, and only those proteins with degree of interactions ≥15 were selected for ease of visualisation. The size of the node indicates degree of interaction with other proteins among the 1124 DEGs. Enriched pathways from pathDIP programme highlighted in colour.
InEx cell phenotype was further explored by assessing secreted factors during resting and stimulated states. InEx cells produced IL-17A, PRF1, granzyme A and GZMB, even under resting conditions. This was congruent with our transcriptomic data. While IL-10 and TNFAIP3 transcripts were observed in InEx cells, production of IL-10 and TNFα at the protein level was limited by unstimulated InEx cells, although they were elevated when stimulated. Additionally, despite limited transcripts, InEx cells produced some IFNγ and granulysin when unstimulated (online supplementary figure S9).
In summary, InEx cells represent a distinctive population of joint-resident CD8+ T cells in AS patients. These cells have a distinct transcriptional profile under both resting and stimulated conditions compared with other joint-derived CD8+ T cells.
Discussion
This study demonstrates how next generation technologies empower examination of the immune system at a previously unattainable level. This approach will be central to understanding AS. Using hierarchal clustering and dimensionality reduction algorithms, we were able to observe differences in immune cell composition and expression of trafficking molecules, most notably in the SF of AS patients.
We identified the InEx mature CD8+ T cell population, with characteristic markers—CD103, β7, CD29, CD49a. These markers were found to be unique to InEx cells among T cell populations, implicating a role in AS pathogenesis. CD103+β7+ expression defines gastrointestinal intraepithelial lymphocytes (IELs)18 with CD49a defining a cytotoxic subset.19 The CD49a+CD29+ integrin heterodimer, known as VLA-1, binds type IV collagen and is commonly found on gut-resident CD8+ T cells.20 Prior studies blocking VLA-1 in immune-mediated inflammation models have confirmed amelioration of gut and joint immune infiltration, synovitis, cartilage damage and clinical disease.21
It can be speculated that InEx cells are involved with apoptosis via ubiquitynation and a complex signalling pathway. In fact, besides restricting NF-κB signalling downstream of tumour necrosis factor receptor 1, A20 (TNFAIP3) regulates multiple ubiquitin-dependent signalling by interacting with E3 enzymes like TRAF6.22 This may be particularly important since TNFAIP3 contains single-nucleotide polymorphisms linked to diseases like RA, psoriasis and IBD.22 A20 also functions as a negative regulator of the inflammasome, which may protect against arthritis.23 Apoptosis could also occur by the granzyme B/perforin cytotoxic pathway24 or granulysin-mediated mechanisms.25 Our expression profiles indicate that InEx cells may reflect a balancing act of cytotoxicity and cytokine release by way of inhibitory (NKG2A, encoded by KLRC1) and activating (NKG2C, encoded by KLRC2) receptors.26 NKG2A has been thought to counterbalance the action of stimulatory NKG2C. In fact, polymorphisms in NKG2A and NKG2C have been reported to be associated with RA.26
InEx cells have the capacity to produce multiple cytokines. Upregulation of IRF4 could suggest a mechanism by which IL-17 and IL-21 production could be observed in InEx cells.27 IRF4 is also known to promote expression and function of Blimp1 and T-bet, transcription factors required for CD8+ T cell differentiation. In fact, impaired IRF4 in peripheral CD8+ T cells severely impacts their antiviral ability, viral clearance and host recovery from influenza infection. Furthermore, IRF4 expression is regulated by TCR signalling through mammalian target of rapamycin (mTOR).28 This is notable in light of our observation of enhanced RPTOR transcripts in InEx cells. Another informative observation was the IL-10 elevation in InEx cells. An anti-inflammatory cytokine, IL-10 has been implicated in RA pathogenesis, particularly due to its increased mRNA levels in RA SFMC.29
It appears that upregulation of CCL5 transcripts could potentially allow InEx cells to recruit myeloid cells such as macrophages, neutrophils and potentially dendritic cells30 to the inflamed synovial microenvironment.
The InEx core transcriptional and phenotypic signature is reminiscent of human tissue-resident memory T cells (TRMs), in their expression of CD103, CD49a, CXCR6 and IL-10, and their lack of CD62L, S1PR1 and CX3CR1.31 Future studies are required to understand InEx cells’ function, TCR clonality and presence in gut as well as joint, which would enable insights into the gut–joint pathway implicated in AS and may lend support to the arthritogenic peptide hypothesis. In fact, CD8+ TRMs have been reported to express high-affinity TCRs,32 an observation which may support our InEx-resident memory theory. Although we have not yet had the opportunity to incorporate gut biopsies in our study, the InEx cells also appear to be quite similar to IELs. This alternative theory might suggest that these cells have a mucosal origin and have potentially trafficked to the joint, which supports the hypothesis of the gut being an initiator of inflammation in AS.
In summary, through unbiased multidimensional analyses, we identified a distinctive trafficking molecule expression pattern in AS joint inflammation. We have also highlighted tissue and disease-preference pattern of marker expression. It is notable that in general CD8+ T cells are heterogeneous and include both cytotoxic and anti-inflammatory features,33 however this has not been previously studied in AS via multiparameter analysis. Our analyses support the concept that immune heterogeneity in T cell populations in AS is far greater than previously appreciated and thus calls for further study. Most importantly, we have identified a novel and potentially pathogenic CD8+CD45RO+ subset, CD103+β7+CD29+CD49a+, that may contribute to AS pathogenesis. The functional capabilities of the InEx versus the NK cells appear distinct, that is, cytotoxic versus cytokine-producing, respectively, even though both share similar trafficking molecule expression. Analysis of target tissues will be informative in future studies, as reflected in the recent finding of elevated CD103, β7, CD49a and CD29 in the colon of human CD8+ T cells.34 Therapeutic approaches using integrin blockers have been successful in IBD, but perhaps need to be better tailored to AS to prove efficacy, given inconsistent results using current antagonists.8 10 11 Current cytokine-blocking agents in AS targeting TNFα or IL-17A have achieved symptomatic improvement but have not reliably prevented radiographic progression in AS nor have proven to be curative.
We acknowledge that our study is limited by sample size and a lack of gut biopsy samples; however, further work involving large sample sizes and tissue-specific data would address these limitations. Additionally, there are some discrepancies in our transcriptomic versus protein analyses, and this may be attributed to a low sample size (which could interfere with statistical analyses) or post-transcriptional modification processes. Notwithstanding, such exploratory approaches and genetic evaluations of TCR sequences and oligoclonality will help pave the way towards understanding of trafficking markers extensively in AS and unravel the complex relationship between the gut and joint axis of inflammation. These findings could prove beneficial for future blocking studies using murine AS models, through which researchers could conclusively determine the role of the identified subpopulation markers. This would establish a causal link and could lay the groundwork for innovative immunotherapy for AS in the future.
Supplemental material
Acknowledgments
At PGCRL, we thank Tina Chen and Daniela Tantalo for help acquiring samples on the CyTOF2 instrument. At PMGC, we thank Neil Winegarden, Julissa Tsao, Vikarna Rampersad and Monika Sharma, who helped process RNA for sequencing and NanoString assays, and Zhibin Lu, who helped with bioinformatics. At TWH, we are grateful to Edwin Speck for help with FACS sorting, to the following clinical research staff at the Spondylitis clinic for assistance in gathering clinical information and patient samples: Sister Esther Paeste, Daeria Lawson, Kevin Seetaram and Ammepa Anton, and to Chiara Pastrello for valued assistance with protein-protein network and pathway enrichment analyses.
References
Footnotes
Handling editor Josef S Smolen
Contributors ZQ, EG and RDI were involved with study conception and design. ZQ and YY processed the blood and synovial fluid samples, while ZQ and EG performed the remainder of the experiments. ZQ, EG and RDI analysed and interpreted the data. ZQ, EG and RDI wrote the manuscript. All authors agreed to publish the data and reviewed the manuscript.
Funding Canadian Institutes of Health Research (CIHR) grant number 159671 Krembil Research Institute Fall 2016 Post-doctoral Fellowship recipient University of Toronto Open Fellowship.
Competing interests None declared.
Patient consent for publication Obtained.
Ethics approval Venous blood and knee synovial fluid extraction obtained under the institutional approval.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.