Article Text

Abstract
Objectives Genetic variations in TNFAIP3 (A20) de-ubiquitinase (DUB) domain increase the risk of systemic lupus erythematosus (SLE) and rheumatoid arthritis. A20 is a negative regulator of NF-κB but the role of its DUB domain and related genetic variants remain unclear. We aimed to study the functional effects of A20 DUB-domain alterations in immune cells and understand its link to SLE pathogenesis.
Methods CRISPR/Cas9 was used to generate human U937 monocytes with A20 DUB-inactivating C103A knock-in (KI) mutation. Whole genome RNA-sequencing was used to identify differentially expressed genes between WT and C103A KI cells. Functional studies were performed in A20 C103A U937 cells and in immune cells from A20 C103A mice and genotyped healthy individuals with A20 DUB polymorphism rs2230926. Neutrophil extracellular trap (NET) formation was addressed ex vivo in neutrophils from A20 C103A mice and SLE-patients with rs2230926.
Results Genetic disruption of A20 DUB domain in human and murine myeloid cells did not give rise to enhanced NF-κB signalling. Instead, cells with C103A mutation or rs2230926 polymorphism presented an upregulated expression of PADI4, an enzyme regulating protein citrullination and NET formation, two key mechanisms in autoimmune pathology. A20 C103A cells exhibited enhanced protein citrullination and extracellular trap formation, which could be suppressed by selective PAD4 inhibition. Moreover, SLE-patients with rs2230926 showed increased NETs and increased frequency of autoantibodies to citrullinated epitopes.
Conclusions We propose that genetic alterations disrupting the A20 DUB domain mediate increased susceptibility to SLE through the upregulation of PADI4 with resultant protein citrullination and extracellular trap formation.
- PAD4
- NET
- peptidyl arginine deiminase
- PADI4
- rs2230926
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
A20 is a well-known negative regulator of NF-κB, and genetic variations in its de-ubiquitinase (DUB) domain, such as rs2230926, are associated with increased risk of rheumatoid arthritis and systemic lupus erythematosus (SLE).
The functional role of genetic alterations in A20 DUB domain is unclear and studies around NF-κB-independent mechanisms are lacking.
What does this study add?
This study reveals a novel functional mechanism of A20 DUB domain disruptions and provides a new genetic link to protein citrullination and extracellular traps in SLE via an upregulation of PADI4.
Our results highlight the importance of protein citrullination and neutrophil extracellular traps in SLE pathogenesis.
How might this impact on clinical practice or future developments?
Our results support a role for PAD4 and protein citrullination in SLE pathogenesis and suggest PAD4 as a potential therapeutic target in diseases associated with A20 DUB-domain alterations.
Introduction
A20 (encoded by TNFAIP3) is a negative regulator of NF-κB-induced expression of proinflammatory and survival genes. Several studies have shown that individuals with the non-synonymous polymorphism rs2230926 located in the A20 de-ubiquitinase (DUB) domain (phenylalanine-to-cysteine change at position 127, F127C), have increased susceptibility to autoimmune diseases, such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA).1–3 Other coding polymorphisms and alternative splicing isoforms affecting the A20 DUB domain are also associated with autoimmune diseases.4 5 DUB-domain polymorphisms are considered to lead to structural changes, reduced DUB activity and increased NF-κB activity,1 4 but recent studies question the role of the A20 DUB domain in NF-κB regulation.6 Although mice with the C103A mutation (cysteine-to-alanine change at position 103 of A20) lack a functional DUB domain,6–9 De et al 6 reported that their innate immune cells show comparable NF-κB-mediated proinflammatory response to wild-type (WT) mice and suggest that the DUB activity of A20 is not necessary for its NF-κB-inhibiting function. Thus, other mechanisms beyond the NF-κB pathway could be responsible for the increased risk of autoimmunity caused by genetic disruption of the A20 DUB domain.
Here, we show that instead of activating the NF-κB pathway, genetic disruptions in A20 DUB domain unexpectedly lead to increased protein citrullination. Protein citrullination is a post-translational modification converting the amino acid arginine into a citrulline on target proteins. This modification is catalysed by a group of Ca2+-dependent enzymes called peptidyl arginine deiminases (PADs).10 Citrullination is a key feature in RA pathogenesis where it is linked to the induction of autoantibodies to citrullinated self-epitopes (reviewed in Catrina et al 11). Additionally, histone citrullination by PAD4 is involved in NET formation, a process whereby neutrophils release chromatin filaments coated with citrullinated histones and antibacterial proteins to trap and kill pathogens.12 13 In SLE, enhanced NET formation has been reported and suggested to contribute the pathogenesis by different mechanisms, such as promoting autoantibody formation to post-translationally modified nuclear antigens,14 15 stimulating release of type I IFNs from plasmacytoid DCs,16 17 and mediating endothelial dysfunction, prothrombotic and atherosclerotic changes.18–21
Here, we explored the functional effects of genetic disruptions in A20 DUB domain using a CRISPR/Cas9-generated human cell line with C103A knock-in (KI) mutation, murine immune cells from A20 C103A mice, and immune cells from healthy individuals and SLE-patients carrying A20 DUB domain polymorphism rs2230926. We show that genetic disruptions in A20 DUB domain cause upregulation of PADI4, leading to increased protein citrullination and extracellular trap formation. Our findings reveal a novel function for A20 DUB domain variants and provides a genetic link emphasising the role of citrullination, extracellular traps and neutrophils as upstream pathogenic mechanisms in SLE.
Methods
SLE-patients and healthy controls
SLE-patients were recruited at the rheumatology clinics in Uppsala, Karolinska (Stockholm), Lund and Linköping University Hospitals and from the four northern-most counties in Sweden. Medical records were reviewed; all patients were examined by a rheumatologist and fulfilled ≥4 American College of Rheumatology 1982 classification for SLE.22 Control individuals were blood donors from Uppsala Bioresource, recruited at the Department of Transfusion Medicine, Uppsala University Hospital, Sweden. The study was approved by the regional ethics review boards of Uppsala, Lund, Linköping, Stockholm and Umeå and all participants gave their informed consent. Demographics of patients and controls are described in online supplementary table S1.
Supplemental material
Patient and public involvement
The research included in this manuscript was conducted and designed without patient or public involvement on study design, outcomes or interpretation of results.
Generation of A20 C103A KI and A20 KO cells by CRISPR/Cas9
Online supplementary figure S1 represents the outline of the generation of C103A KI and A20 KO cells. Targeted KI mutation C103A was generated by TG-to-GC substitution in human TNFAIP3 DUB domain (replacing cysteine at position 103 for alanine) using CRISPR/Cas9 in U937 cells. Guide RNAs were designed using the ZiFiT software (http://zifit.partners.org/ZiFiT/) and cloned into a pCas9-GFP expression vector. C103A cell clones were selected using green fluorescent protein (GFP) expression and restriction fragment length polymorphism and confirmed with sanger sequencing and droplet digital PCR. Three successfully generated homozygous C103A KI clones (C103A KI1-KI3) were confirmed deriving from two different guide RNAs to account for possible off-target effects. Control cells underwent the same processes as the targeted cells but in absence of plasmids. A20 knock-out (KO) cells were generated by transfecting U937 cells with the Cas9 expression vector coding the same guide RNAs as previously described, but in the absence of targeting vector and were screened for the absence of A20 protein by western blot.
Neutrophil isolation
Blood from SLE-patients was collected in vacutainer tubes with heparin (BD Bioscience) and neutrophils were separated using density gradient centrifugation in LymphoPrep (StemCell Technologies) after erythrocyte sedimentation in 2% Dextran (Sigma). Purity was >95% as assessed by Sysmex XT 1800i (Sysmex Kobe, Japan). Murine neutrophils were isolated from whole blood from 8 to 12-week-old sex-matched WT and A20 C103A KI mice using EasySep Mouse Neutrophil Enrichment Kit (Stemcell Technologies) after erythrocyte lysis with BD Pharm Lyse lysing solution (BD Bioscience). Blood from 3 to 5 mice were pooled together in each experiment to obtain enough neutrophils. Experiments with human and murine neutrophils were performed in RPMI with 10% FBS (Gibco).
NET induction and immunofluorescence staining
Equal number of neutrophils were plated in eight-well chamber glass slides (Lab-Tek II Chamber Slide, NUNC) precoated with Poly-L-lysine (Sigma-Aldrich). PAD4 inhibitor GSK484 (10 µM) or vehicle (dimethyl sulfoxide (DMSO)) were added to the cells 30 min before stimulation with 1 or 4 µM Ionomycin +2 mM CaCl2. After 1 hour and 45 min for murine neutrophils, or after 1 hour for human neutrophils, cells were fixed in 4% paraformaldehyde (VWR Chemicals), permeabilised with 0.25% Triton X-100 (Sigma), and blocked in 1% BSA/2% goat serum. Cells were stained with antibodies against citrullinated histone H3 (ab5103; Abcam) and Myeloperoxidase (clone 2C7, ab25989; Abcam) followed by Goat anti-Rabbit IgG (H+L, Alexa Fluor 568; Thermo Fisher Scientific) and Goat anti-Mouse IgG (H+L, Alexa Fluor 633; Thermo Fisher Scientific) secondary antibodies. Nuclei and DNA were visualised by Hoechst (Life technologies) and SYTOX Green Nucleic Acid Stain (Thermo Fisher Scientific). Fluorescence microscopy was performed using Carl Zeiss LSM 880 confocal microscope and images analysed by ZEN v.2.3 software. See online supplementary figure S9C-D for control stainings. Researchers were blinded to the patient genotype when performing experiments and analysis.
Detection of secreted citrullinated histone H3 and dsDNA
The supernatant from U937 cells and primary neutrophils was collected after 6 or 1 hour after Ionomycin treatment, respectively. Double-stranded DNA (dsDNA) was measured using Quant-iT PicoGreen dsDNA reagent (Invitrogen). For detection of citrullinated histone H3, the proteins in the supernatant were precipitated with 20% trichloroacetic acid (Sigma-Aldrich), resuspended in 2,5x LDS loading buffer (Invitrogen) and analysed by western blotting.
Statistical analyses
All data are represented as mean+SD unless otherwise indicated. Figure legends specify the type of statistical test used in different experiments. Graphpad Prism (v7) was used for statistical analyses of in vitro data. One-tailed comparisons were used when confirming a previously observed effect, otherwise two-tailed comparisons were performed. Pairwise comparison was used if inter-experimental baseline signals differed substantially, and is then specified in the figure legend. For statistical analyses of large data sets, statistical details are described in corresponding method sections. P values below 0.05 were considered significant.
Detailed information for all methods can be found in online supplementary material.
Supplemental material
Results
A20 DUB-domain disruption does not increase NF-κB activation
To address the functional effect of A20 DUB-disruption in development of autoimmunity, we used CRISPR/Cas9 genome-editing to introduce the DUB-inactivating C103A mutation into the human U937 monocytic cell line (C103A KI cells, figure 1A and online supplementary figure S1). We performed whole transcriptome analysis in WT, C103A KI and A20 KO U937 cells. When we analysed the expression of known NF-κB target genes23 by Gene Set Enrichment Analysis, we found that while a complete A20 KO caused a significant increase in NF-κB target gene expression on LPS-stimulation, C103A KI cells did not demonstrate enhanced NF-κB signalling (figure 1B). Moreover, we observed no alteration in TLR-induced NF-κB signalling in bone marrow derived macrophages (BMDMs) from C103A KI mice (online supplementary figure S2), supporting published data indicating that the C-terminal zinc fingers, and not the DUB domain, are involved in NF-κB signalling.6 24

Genetic disruption of A20 de-ubiquitinase (DUB) domain does not lead to enhanced NF-ĸB activation. (A) Generation of A20 C103A knock-in (KI) mutation in U-937 cell line using CRISPR/Cas9. Three different homozygote C103A KI clones were generated and Sanger sequencing of the homozygote clones is shown to the right. (B) RNA sequencing data from lipopolysaccharide (LPS)-stimulated human U-937 cells (A20 WT, knock-out (KO) and C103A KI) were analysed using gene set enrichment analysis (GSEA). NF-ĸB target genes were significantly enriched in A20 KO cells (left), but not in A20 C103A KI cells (right), when compared with their wild-type counterparts. (C) Peripheral blood mononuclear cells (PBMCs) from individuals with A20 DUB polymorphism rs2230926 (n=10, CA or CC) or without (n=7, AA), were treated with LPS for 6 hours and intracellular staining of tumour necrosis factor-alpha and IL-6 was measured by flow cytometry in monocytes (CD14 Mo and CD16 Mo) and myeloid dentritic cells (mDC). No significant differences were found between genotypes using a two-tailed t-test. CA represents individuals that are heterozygous for rs2230926 risk polymorphism, CC are homozygous and AA represents individuals without rs2230926 risk allele. FDR, false discovery rate; NES, normalised enrichment score; OTU, ovarian tumour domain; ZF, zinc finger.
Next, we asked whether the A20 DUB-domain polymorphism rs2230926 interferes with NF-κB activation. We genotyped 982 SLE-patients and 1980 healthy individuals from a Swedish SLE cohort (online supplementary table S1) and in agreement with earlier studies,1 3 we found that rs2230926 associated with SLE (table 1, OR=1.83, p=1×10− 6). Additionally, we found it being an even stronger risk factor for SLE in males (OR=3.26, p=1.27×10−5). We used flow cytometry to measure the levels of IL-6 and tumour necrosis factorα, two NF-κB-induced cytokines regulated by A20, in LPS-stimulated monocytes and myeloid dendritic cells from healthy individuals with rs2230926 risk variant (CA or CC) or without (AA). However, no difference could be observed between genotypes (figure 1C and online supplementary figure S3). The levels of A20 were similar in both groups (online supplementary figure S4). Together, these results indicate that the consequences of A20 DUB-domain genetic variants associated with autoimmunity are not mediated via enhanced NF-κB signalling.
Association between rs2230926 and SLE
Genetic A20 DUB variants express elevated PADI4 levels
To explore other possible mechanisms regulated by the A20 DUB-domain mutations, beyond NF-κB signalling, we compared the global gene expression profiles between A20 KO, C103A KI and WT U937-cells. We observed that unstimulated C103A KI cells presented a large number of differentially expressed genes compared with WT cells and KO cells (online supplementary table S2). One of the top hits of differentially expressed genes in C103A KI cells was PADI4, encoding PAD 4 (PAD4), while no PADI4 upregulation was observed in A20 KO cells (online supplementary table S2). qPCR analysis showed around 30-fold PADI4 upregulation in all C103A KI clones when compared with WT clones (figure 2A). This increased expression was also reflected at the protein level (figure 2B). PADI4 was also significantly elevated in C103A mouse BMDMs compared with WT, indicating that this effect is not restricted to the genetically modified U937-cells (figure 2C). We then analysed PADI4 expression in human PBMCs from healthy individuals with or without rs2230926 risk allele, and found significantly increased PADI4 levels in the subjects with rs2230926 risk allele (figure 2D). The difference in PADI4 expression was confirmed in PBMC-derived monocytes and NK cells (figure 2E and online supplementary figure S5). PAD4 protein levels were measured by western blot in isolated monocytes and were also significantly higher in individuals with the rs2230926 risk allele (figure 2E and online supplementary figure S6).

A20 de-ubiquitinase (DUB) domain disruption leads to PADI4 over expression. (A) Confirmation of RNAseq data using qPCR analysis of the expression of PADI4 mRNA in U937 wild-type (WT) (n=4), A20 knock-out (KO) (n=4) and A20 C103A knock-in (KI) cells (n=3). Individual squares or circles represent individual CRISPR/Cas9-generated cell clones. (B) PAD4 protein expression measured by western blot in DMSO-treated individual U937 WT and C103A KI clones. (C) qPCR analysis of the expression of murine PADI4 mRNA in BMDMs from WT and A20 C103A KI mice (n=3 per group). (D) qPCR analysis of the expression of PADI4 mRNA in human PBMCs (n=13 AA, n=13 CA). (E) qPCR analysis of the expression of PADI4 in PBMC-derived human monocytes (left graph, n=9 AA, n=13 CA). The right graph represent Western blot quantification of PAD4 protein relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in PBMC-derived human monocytes (n=7 AA, n=6 CA). CA represents individuals that are heterozygous for rs2230926 risk polymorphism and AA represents individuals without rs2230926 risk allele. (A,C,D,E) PADI4 expression levels are normalised to house-keeping genes GUSB and ACTB and are represented as relative mRNA expression to average PADI4 expression in controls (WT or AA). Statistical differences between groups were calculated using two-tailed t-test with Welch correction (D) or non-parametric one-tailed Mann-Whitney test (A,C,E). Bars represent mean+SD. *p<0.05, **p<0.01. BMDM, bone marrow derived macrophage.
A20 DUB-domain disruption enhances protein citrullination and NET formation
Given the strong link between citrullination and autoimmune pathologies, we were encouraged to explore the functional consequences of PADI4 over expression caused by A20 DUB mutations. Flow-cytometric analysis with F95 pan-citrulline antibody showed increased protein citrullination in the C103A KI cells on PAD4 activation with Ionomycin and CaCl2 when compared with WT cells (figure 3A). Protein citrullination in cells with mutated A20 is dependent on PAD4, as specific PAD4 inhibition with small-molecule inhibitor GSK48412 decreased citrullination (figure 3B, online supplementary figure S7). Next, we explored the role of A20 DUB variants on histone H3 citrullination (H3cit), NET formation and release of nuclear antigens linked to the pathogenesis of SLE. As extracellular trap formation is not restricted to neutrophils but is also seen in monocytes and macrophages,25 26 we used the U937 cells to study these processes. We observed an increased cytoplasmic H3cit expression in the C103A KI cells while no differences were seen in nuclear levels of citrullinated histones between C103A KI and WT cells (online supplementary figure S8). As H3cit and dsDNA are released in the NET formation, we measured H3cit and dsDNA in the supernatant of Ionomycin-treated U937 cells. C103A KI cells demonstrated increased levels of both H3cit and dsDNA on Ionomycin-treatment compared with the WT cells, and the levels decreased significantly on PAD4 inhibition (figure 3C–F). A trend to increased spontaneous dsDNA release was also seen in PBMCs from individuals with the rs2230926 risk allele (figure 3G).

Disruption of A20 de-ubiquitinase (DUB) domain is associated with PAD4-dependent increase in protein citrullination and release of nuclear antigens. (A–B) Total intracellular protein citrullination was measured in wild-type (WT) and C103A knock-in (KI) U937 cells using flow cytometry after stimulation with ionomycin. The effect of PAD4 inhibitor GSK484 on protein citrullination was assessed in U937 A20 C103A KI cells (B). Data points represent experimental duplicates in three independent experiments. (C) Western blot performed on supernatants from 1 million WT or A20 C103A KI U937 cells stimulated with ionomycin for 6 hours, with or without GSK484. GAPDH expression from the cell lysate is represented as an input control. representative blot from one out of four experiments. (D, E) Quantification of H3cit Western blot signal from four independent experiments is represented as mean H3cit density +SD. (F) the presence of cell free double stranded DNA (dsDNA) in the supernatants of U937 WT and KI cells were measured after 6 hours of ionomycin treatment. Results from four independent experiments are represented as mean % increase compared with unstimulated cells +SD. (G) The presence of dsDNA in the supernatants of unstimulated PBMCs derived from healthy individuals with the rs2230926 risk variant (CA) or without (AA). Statistical differences between groups were performed using paired two-tailed t-test (D–F), two-tailed t-test (G) or paired two-tailed non-parametric Wilcoxon test (A,B). Unless different concentrations are stated, 4 µM ionomycin was used in the presence of 2 mM CaCl2. GSK484 was used at 10 µM. *p<0.05, **p<0.01, ***p<0.001.
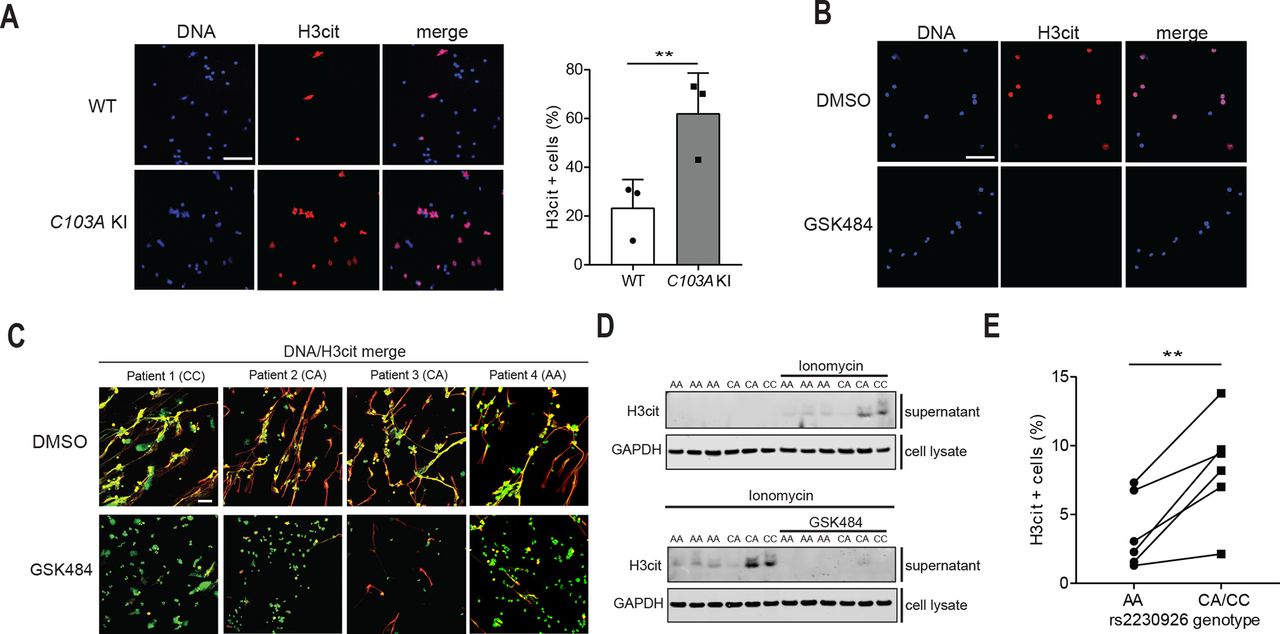
Additionally, we studied Ionomycin-induced NET formation in neutrophils ex vivo, and quantified the number of H3cit-positive cells as a marker of NETs, as previously described.12 27 28 Ionomycin-treatment of blood-derived murine neutrophils led to NET generation characterised by increased H3cit expression and DNA spreading with H3cit/DNA co-localisation (online supplementary figure S9A). A higher percentage of H3cit-positive neutrophils was found in C103A KI mice than in WT mice, linking A20 DUB-domain disruption to increased NET formation (figure 4A). Inhibition of PAD4 in murine neutrophils led to reduction of Ionomycin-induced NETs (figure 4B). Human neutrophils stimulated with Ionomycin also formed NETs characterised by H3cit expression which co-localise with DNA and myeloperoxidase (online supplementary figure S9B). We next studied NET formation in neutrophils isolated from 12 genotyped SLE patients (six with the rs2230926 risk allele and six without). When stimulated with high-dose (4 µM) Ionomycin, most of the neutrophils underwent full-blown NET-formation with extruding DNA chromatin fibres and H3cit expression (figure 4C). Ionomycin-induced NETs and release of H3cit could be prevented by PAD4 inhibition, confirming the central role of PAD4 in this process (figure 4C,D). In order to get a window for NET quantification, we used a lower dose of Ionomycin (1 uM) and quantified the number of H3cit-expressing neutrophils (figure 4E shows quantification and online supplementary figure S9E shows example images). Neutrophils from SLE-patients with rs2230926 risk allele expressed a higher percentage of H3cit positive cells than SLE-patients without risk allele. The higher propensity of NET formation in SLE-patients with rs2230926 was supported by increased number of neutrophils showing DNA extrusions and increased amount of secreted cell-free DNA in the neutrophil supernatants (online supplementary figure S10). Ionomycin-induced DNA extrusions in SLE-derived neutrophils were further confirmed to show characteristic co-expression of DNA and myeloperoxidase (online supplementary figure S11).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disruption of A20 de-ubiquitinase (DUB) domain is associated with PAD4-dependent increase in NET formation. (A–C) Immunostaining and confocal microscopy of citrullinated H3 (H3cit, red) and DNA (SytoxGreen in green or Hoechst in blue) in murine and human neutrophils. Scale bar=50 µm. (A) Comparison of H3cit expression in Ionomycin-stimulated neutrophils from wild-type (WT) and C103A Ki mice. Results are expressed as mean percentage H3cit-postitive cells compared with total cell number +SD, and are pooled values from three independent experiments including totally 13 WT and 11 C103A KI mice. Each data point represents the mean of one experiment. (B) Neutrophils from C103A KI mice were pretreated with PAD4 inhibitor GSK484 or vehicle (DMSO) before NET-induction by ionomycin. Representative image. (C–E) Neutrophils from human systemic lupus erythematosus (SLE)-patients that were homozygous (CC, n=1),heterozygous (CA, n=5), or lacked (AA, n=6) rs2230926, were induced to form NETs in vitro. (C,D) Human SLE-derived neutrophils were pretreated with GSK484 or vehicle (DMSO) and NET formation was induced with 4 µM ionomycin. Representative images show reduced H3cit expression in GSK484-treated cells (C). H3cit was released in the supernatant on ionomycin stimulation of SLE-neutrophils and could be blocked by PAD4 inhibition (D). (E) Neutrophils from SLE-patients were treated with 1 µM ionomycin and the number of H3cit-positive cells was counted and represented as percentage compared with total cell number. Samples analysed on the same day are connected with a line. Statistical differences between groups were performed using a two-tailed t-test (A) or paired two-tailed t-test (based on age-matched patients from the same experiment) (H). Unless different concentrations are stated, 4 µM ionomycin was used in the presence of 2 mM CaCl2. GSK484 was used at 10 µM. *p<0.05, **p<0.01.
A20 rs2230926 associates with presence of antibodies to citrullinated antigens in SLE
The presence of antibodies against citrullinated proteins (ACPA) is a hallmark of RA, but ACPA can also be detected in a small group of SLE-patients.29 Thus, we analysed antibodies to cyclic citrullinated peptides (CCP) in 195 SLE patients in our cohort, and found a significantly increased frequency of IgG anti-CCP in the patients with rs2230926 allele when compared with the rest of the patients (24% vs 4%, table 2). This finding further indicates that A20 DUB-domain variant is strongly associated with protein citrullination and modification of immunogens in humans. A weak association with the occurrence of malar rash and seizures and the presence of rs2230926 allele was observed, whereas no other clinical associations were found (online supplementary table S4).
Serum autoantibodies in SLE-patients
Discussion
In this study, we reveal a novel functional mechanism for the autoimmunity risk mediated by the genetic disruption of A20 DUB-domain, that is, increased protein citrullination and extracellular trap formation due to upregulation of PADI4. Even though polymorphisms in A20 DUB region are among the most frequently reported risk alleles in RA and SLE, their functional impact has not been well established. Several studies have attributed an NF-κB-regulating role to the A20 DUB domain.4 7 8 However, there is a lack of functional studies in human cells and the mechanistic data describing the role of A20 DUB activity are mainly performed in cell-free in vitro systems.7 8 Our data show that A20 DUB activity is not necessary for NF-κB inhibition in human immune cells and is in agreement with a recent report by De et al 6 using immune cells from A20 C103A mice. Our data does not exclude however, that other reported polymorphisms in the gene of A20, such as those leading to decreased A20 protein levels or those affecting the A20 zinc fingers, impact NF-κB activation.30 31
Increased NET formation have been described in SLE patients previously and has been connected both to autophagy and to a defective clearance of NETs.32 33 Although histone H3 citrullination (H3cit), NET formation and release of nuclear antigens have been linked to the pathogenesis of SLE, the role of these mechanisms as drivers of disease phenotypes has not been clear. Our observations revealing a novel genetic link between SLE risk and protein citrullination and NET formation, provide support for this pathway as a disease-driving mechanism in SLE. These findings are consistent with studies showing that PAD inhibition is immunomodulatory in the murine NZM lupus model characterised by Type I IFNs, vascular dysfunction and prothrombotic risk, suggesting PAD-mediated NET formation as an upstream modulator of the various disease phenotypes.34 Furthermore, blocking mitochondrial ROS production and subsequent spontaneous NET formation in a mouse model of lupus resulted in reduced disease severity.35 Admittedly, the role of NETs is challenged in other experimental lupus-like models, such as the MRL. Fas-lpr or pristane-induced models, where PADI4 deletion or Nox2 deletion (disrupting NET formation) failed to ameliorate the phenotype.36–38
An outstanding question still is how the mutations in the A20 DUB domain can regulate PADI4. Mass spectrometry data of proteins bound to the different A20 forms (WT, C103A and F127C) did not reveal any direct binding between A20 or any known or predicted transcription factors regulating PADI4 (data not shown), which may indicate that a multistep mechanism is involved. The lack of PADI4 upregulation in A20 KO cells and the fact that heterozygote individuals for rs2230926 present increased PADI4 levels and increased autoimmunity risk, suggest a dominant acquired novel function of the mutated A20. Additional studies are thus warranted to establish the full mechanism clarifying how mutated A20 DUB domain regulates PADI4 expression.
Our findings, which were unexpected, reveal a novel genetic alteration involved in the regulation of protein citrullination, proposing an explanation for the contribution of A20 DUB-domain polymorphisms in the autoimmune pathogenesis. The results presented in this study highlight protein citrullination, extracellular trap formation and neutrophils as key factors in the pathogenic process in SLE and further support PAD4 as a target for the prevention and treatment of this disease.
References
Footnotes
Handling editor Josef S Smolen
ZJ and RR contributed equally.
Contributors LO, ZR, RR, MR, JM, KT, SJ, AK, SN, SA planned, performed and analysed experiments. LÖ and PJ performed bioinformatic analyses. DL performed data analysis and AA performed statistical analyses. PC and LFY were involved in data discussion and supervision of study design experiments. JKS and A-CS generated and provided genotype data. ES, AJ, IG, KT, AK, SR-D and AB collected and provided patient and/or control material and clinical characterisation. M-LE planned experiments and provided patient material. CS provided scientific and experimental input and patient material. LR planned experiments, provided patient material and clinical data and was involved in data discussion and manuscript writing. BC was involved in study design, data discussion and supervision of experiments. OV designed experiments, analysed data and supervised the study. OV, LO, BC and LR wrote the manuscript. All authors critically reviewed and approved the manuscript.
Funding The study was funded by DISSECT AstraZeneca-SciLifeLab collaboration, The Swedish Research Council, Knut and Alice Wallenberg Foundation, Swedish Rheumatism Foundation, King Gustaf V’s 80-year Foundation, Swedish Society of Medicine, the Ingegerd Johansson donation, and Medical Research Council Programme Grant MRC000985.
Competing interests LO, ZR, RR, LÖ, MR, LFY, SJ, JM, KT, PJ, BC and OV were employees at AstraZeneca Group while performing this study and may have stock/stock options in AstraZeneca. AB received research grant from DISSECT, partly funded by AstraZeneca. LR reports grants and personal fees from Astra Zeneca during the conduct of the study and personal fees from Biogen outside the submitted work. AstraZeneca provided funding to DISSECT for the conduct of this study. The remaining authors declare that they have no competing interests. There are no patents involved.
Patient consent for publication Not required.
Ethics approval The study was approved by the regional ethics review boards of Uppsala, Lund, Linköping (No. M75-08/2008), Stockholm and Umeå. Use of tissue for ex vivo functional and mechanistic studies (bone marrow and blood) were approved by the local ethics committee in Gothenburg.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available in a public, open access repository. Data are available upon reasonable request.