Article Text

Abstract
Objective To evaluate the efficacy and safety of tocilizumab, an interleukin-6 receptor antibody, in patients with adult-onset Still’s disease.
Methods In this double-blind, randomised, placebo-controlled phase III trial, 27 patients with adult-onset Still’s disease refractory to glucocorticoids were randomised to tocilizumab at a dose of 8 mg/kg or placebo given intravenously every 2 weeks during the 12-week, double-blind phase. Patients received open-label tocilizumab for 40 weeks subsequently. The primary outcome was American College of Rheumatology (ACR) 50 response at week 4. The secondary outcomes included ACR 20/50/70, systemic feature score, glucocorticoid dose and adverse events at each point.
Results In the full analysis set, ACR50 response at week 4 was achieved in 61.5% (95% CI 31.6 to 86.1) in the tocilizumab group and 30.8% (95% CI 9.1 to 61.4) in the placebo group (p=0.24). The least squares means for change in systemic feature score at week 12 were –4.1 in the tocilizumab group and –2.3 in the placebo group (p=0.003). The dose of glucocorticoids at week 12 decreased by 46.2% in the tocilizumab group and 21.0% in the placebo group (p=0.017). At week 52, the rates of ACR20, ACR50 and ACR70 were 84.6%, 84.6% and 61.5%, respectively, in both groups. Serious adverse events in all participants who received one dose of tocilizumab were infections, aseptic necrosis in the hips, exacerbation of adult-onset Still’s disease, drug eruption and anaphylactic shock.
Conclusion The study suggests that tocilizumab is effective in adult-onset Still’s disease, although the primary endpoint was not met and solid conclusion was not drawn.
- adult-onset Still’s disease
- tocilizumab
- randomised
- efficacy
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Suppression of pro-inflammatory cytokines has been reported to be effective in many case reports with adult-onset Still's disease, however, there has been no placebo-controlled trial on these drugs.
What does this study add?
This is the first double-blinded, randomised placebo-controlled trial conducted in patients with adult-onset Still's disease that suggests tocilizumab, an anti-interleukin-6 receptor monoclonal antibody, is effective in improving systemic symptoms and decreasing glucocorticoid dose.
How might this impact on clinical practice or future developments?
Our findings provide useful insights in the management of adult-onset Still’s disease and in designing and conducting future clinical trials on adult-onset Still’s disease.
Introduction
Adult-onset Still’s disease is a rare, systemic inflammatory disorder of unknown aetiology characterised by high spiking fever, evanescent rash and polyarthritis.1 In addition to these major symptoms, other features simultaneously occur with multiple organ involvement, including sore throat, lymphadenopathy, hepatosplenomegaly, and elevated serum liver enzymes and ferritin. Glucocorticoids are the first-line treatment for this disease, and the initial response to glucocorticoids is generally good despite intensive systemic inflammation. Overall treatment, however, remains challenging because high-dose glucocorticoids sometimes fail to cause remission, with occasionally fatal consequences, and dependence on glucocorticoids is frequently observed with a relapse of symptoms along with dose tapering or discontinuation, which leads to organ damage accrual and long-term side effects.2 3
Immunosuppressive agents, such as methotrexate and ciclosporin, have been used as a steroid-sparing drug, but their effectiveness is limited.4 5 Progress in the understanding of the critical role of proinflammatory cytokines in the pathogenesis of adult-onset Still’s disease has led to pilot use of anticytokine agents, resulting in an increasing number of successful case reports in patients who were unresponsive to conventional treatments.6–10 Furthermore, systemic juvenile idiopathic arthritis, previously known as Still’s disease, has responded significantly better to canakinumab, an anti-interleukin-1β monoclonal antibody, and tocilizumab, an anti-interleukin-6 receptor monoclonal antibody, than to placebo in several randomised controlled trials.11–16 Since adult-onset Still’s disease closely resembles systemic juvenile idiopathic arthritis in terms of pathogenesis and cytokine profiles, these anticytokine inhibitors are promising treatments for adult-onset Still’s disease. However, there have been no randomised, placebo-controlled trials on these anti-interleukin-1β or anti-interleukin-6 treatments in patients with adult-onset Still’s disease.
This phase III trial was conducted to evaluate the efficacy and safety of tocilizumab in refractory adult-onset Still’s disease. This article reports the results from the 1-year, multicentre, randomised, double-blind, placebo-controlled trial.
Subjects and methods
Study design
This multicentre, randomised, double-blind, placebo-controlled phase III trial was conducted at eight centres in Japan as an investigator-initiated phase III trial and consisted of three parts: part 1, a randomised, double-blind, placebo-controlled, 4-week phase with a fixed dose of glucocorticoids; part 2, continued double-blind, placebo-controlled, 8-week phase with a conditional decrease in glucocorticoid dose; and part 3, a single, open-label, 40-week phase. This study is registered with UMIN (trial registration number: UMIN000012987).
Patients were recruited from 31 January 2014 to 31 July 2016 and randomly assigned (1:1) by a central automatic system to receive intravenous tocilizumab at a dose of 8 mg/kg or placebo every 2 weeks during parts 1 and 2. All patients received intravenous tocilizumab at the same dose and interval during part 3. The tocilizumab dose was determined in reference to trials in patients with juvenile idiopathic arthritis.13 14 Patients and all medical staff, including doctors, nurses and pharmacists, were unaware of the identity of the trial drugs at all times. Use of concomitant disease-modifying antirheumatic drugs or immunosuppressive agents was prohibited during the study. Doses of glucocorticoids were tapered during parts 2 and 3 according to the predefined rules when patients met the American College of Rheumatology (ACR) 50 criteria without fever: glucocorticoid doses equivalent to prednisolone at least 30 mg/day were tapered by 5–10 mg/day every week, 15–29 mg/day prednisolone by 2.5–5 mg/day every 2 weeks, 7.5–14 mg/day prednisolone by 1.5–2.5 mg/day every 4 weeks, and 5–7.4 mg/day prednisolone by 0.5–1.5 mg/day every 4 weeks. Tapering to prednisolone less than 5 mg/day was at the discretion of the investigators. Glucocorticoids were increased by about 50% within the initial dose (the exact dose was at the investigator’s discretion) when patients did not meet the ACR50 criteria without fever on two consecutive visits. Patients were omitted from the rest of part 2 and were entered into part 3 (escape) when they did not meet the criteria for ACR20 achievement without fever at the beginning of part 2 or ACR50 achievement without fever under the initial dose of glucocorticoids during part 2.
In part 3, the interval of tocilizumab administration could be prolonged in patients who could reduce their glucocorticoids equivalent to or less than 5 mg/day of prednisolone and maintained in stable disease for 4 weeks or more. The prolongation of the interval should be conducted by 1 week. An interval of more than 5 weeks was not allowed. The interval of tocilizumab administration could be shortened to every week at the discretion of the investigators if the response to tocilizumab was considered insufficient. The dosing interval was determined in reference to the trial in juvenile idiopathic arthritis.13
Patients
Eligible patients were 20 years or older with disease onset at more than 15 years old who had been diagnosed with adult-onset Still’s disease with Yamaguchi criteria,17 and had shown inadequate response to more than 2 weeks of glucocorticoids equivalent to at least 0.5 mg/kg/day of prednisolone, with a stable dose of current glucocorticoids equivalent to 10 mg/day of prednisolone for at least 2 weeks. Patients had at least two swollen joints and two tender joints and had ≥1 clinical point of systemic feature score (at least one of the following should be present: fever, rash, lymphadenopathy, hepatosplenomegaly or serositis).18 Patients’ blood tests showed erythrocyte sedimentation rate of at least 20 mm/hour or C reactive protein of at least 1.0 mg/dL.
For 2 weeks before the study drug was first given, treatment with immunosuppressive drugs and disease-modifying antirheumatic drugs was prohibited. Treatment with infliximab, golimumab, adalimumab, abatacept, certolizumab-pegol and leflunomide was not allowed for 12 weeks and etanercept for 4 weeks before patients were given the study drug. Patients were excluded if they had important concurrent medical or surgical disorders, including leucocytopaenia (<3.0×109/L), neutrocytopaenia (<1.0×109/L), lymphocytopaenia (<0.5×109/L) or thrombocytopaenia (<50×109/L), elevated alanine transaminase (>5 times of an upper limit of each site), elevated total bilirubin (>3 times of an upper limit of each site), serious disease assessed by investigators as inappropriate and a history of malignancy within 5 years. All patients were examined for active infections, especially viral hepatitis and tuberculosis.
Assessments and outcomes
Baseline information included age, sex, disease duration, body weight and immunological profile. Clinical and laboratory assessments were done, including fever, skin rash, lymphadenopathy, hepatosplenomegaly, serositis, number of swollen joints and tender joints, patient’s global assessment using a 100 mm Visual Analogue Scale, patient’s pain using Visual Analogue Scale, physician’s global assessment using Visual Analogue Scale, physical function using the Health Assessment Questionnaire Disability Index, complete blood cell counts, erythrocyte sedimentation rate, C reactive protein, liver enzymes and ferritin, every 2 weeks in parts 1 and 2 and every 4 weeks in part 3. The ACR core set and systemic feature score were evaluated.18 19 ACR20, ACR50 and ACR70 responses were defined as both improvement of 20%, 50% and 70% in the number of tender and number of swollen joints and 20%, 50% and 70% improvement in at least three of the remaining five variables of the ACR core set, respectively. Systemic feature score consisted of five clinical and five laboratory assessments. Clinical features included fever, rash, lymphadenopathy, hepatosplenomegaly and serositis, and the laboratory features were erythrocyte sedimentation rate, C reactive protein, leucocyte count, haemoglobin level and platelet count. Each clinical feature was assigned a score of 1 (present) or 0 (absent). At baseline, each laboratory feature was assigned a score of 1 when erythrocyte sedimentation rate was ≥20 mm/hour, C reactive protein was ≥1.0 mg/dL, white cell count was ≥12×103/μL, haemoglobin was ≤11 g/dL and platelet count was ≥400×103/μL. At follow-up visits, laboratory parameters were scored as follows: for erythrocyte sedimentation rate, 0 if <20 mm/hour or if decreased by ≥30% compared with baseline, or 1 if it increased or decreased by <30% compared with baseline; for C reactive protein, 0 if <1.0 mg/L or if decreased by ≥30% compared with baseline, or 1 if it increased or decreased by <30%; for white cell count, 0 if ≥12×103/μL or if decreased by 20% compared with baseline, or 1 if it increased or decreased by <20%; for haemoglobin level, 0 if ≥11 g/dL or if increased by ≥20% compared with baseline, or 1 if it decreased or increased by <20%; and for platelet count, 0 if ≥400×103/μL or if decreased by ≥20% compared with baseline, or 1 if it increased or decreased by <20%.
One of the two coprimary outcomes was the proportion of patients who achieved ACR50 response at week 4, and the other primary outcome was the proportion of patients who had an ACR50 response at week 12 (the primary time point was week 4). We recognised that 4 weeks would be too short for a primary outcome, but at the same time had a great concern about a risk for critically uncontrollable exacerbation of adult-onset Still’s disease in the placebo group. Therefore, we predefined that the primary time point of the primary outcomes was week 4. We selected ACR50 as a primary outcome measure instead of ACR20 in reference to the results from the trial in patients with juvenile idiopathic arthritis,16 in which JIA ACR50 response showed greater effect size than juvenile idiopathic arthritis ACR30. The secondary endpoints were changes in glucocorticoid dose, ACR20, ACR50, ACR70, systemic feature score, and presence of fever and skin rash at each time point. Patients were monitored for safety throughout the 52-week period.
Statistical analyses
In the protocol, sample size calculation was based on the first of the coprimary endpoints, the proportion of patients who achieved ACR50 at week 4. The ACR50 response rate was assumed to be 65% in the tocilizumab group and 15% in the placebo group, in reference to the trials in patients with juvenile idiopathic arthritis.13 14 Under these assumptions, 17 patients were needed in each group to have power of 80% with a two-tailed significance level of 5%. During the clinical trial, we noted that recruitment was proceeding at a slower pace than anticipated because of the difficulty in the enrolment of eligible patients. In May 2016, we performed a blind review to assess whether a reduction in the number of patients was possible. The calculation predicted an ACR50 response in the tocilizumab group to be 80%, assuming that ACR50 in the placebo group was 15%, suggesting that 12 patients per group yielded a power of 80%. We had to accept the risk that our trial would fail to demonstrate significance in the primary outcome despite a true difference, and decided to close recruitment after at least 24 patients were enrolled.
We used descriptive statistics to summarise demographic and baseline disease characteristics by treatment group. The primary analysis population for efficacy evaluation in the double-blind phase (parts 1 and 2) was full analysis set, which included patients who received at least one dose of the study medication. All analyses, including those for the secondary endpoints, were prespecified in the statistical analysis plan before unblinding. The coprimary outcomes in this trial were the ACR50 response rates at weeks 4 and 12 with a predefinition that a test for week 12 would only be evaluated if between-group comparison at week 4 was significant with an alpha=0.05 (two-tailed). We used Fisher’s exact test for dichotomous response outcomes; patients were categorised as either having a response or not having a response. Patients who did not complete the study, escaped from part 2 to part 3, or received weekly intravenous tocilizumab because of inadequate response to biweekly tocilizumab were classified as a non-responder. We used the Clopper-Pearson method to calculate the 95% CIs of proportions. For repeated measurements in part 1, we used mixed-effects model for repeated measurements with unstructured covariance matrix to compare the mean change from baseline between the two groups. For outcomes based on changes from baseline, we used an analysis of covariance model, which contains treatment group as a factor and baseline data as a covariate. The least squares means for the change from baseline were estimated by treatment group with the analysis of covariance model. For missing data in parts 2 and 3, we used the last observation carried forward method. We used the Kenward-Roger method to adjust the df for error. Safety data included full-exposure data for each patient. We defined serious adverse events in accordance with the definition in the International Conference on Harmonisation guidelines.20 We used SAS V.9.4 for all analyses.
Results
Patient flow and characteristics
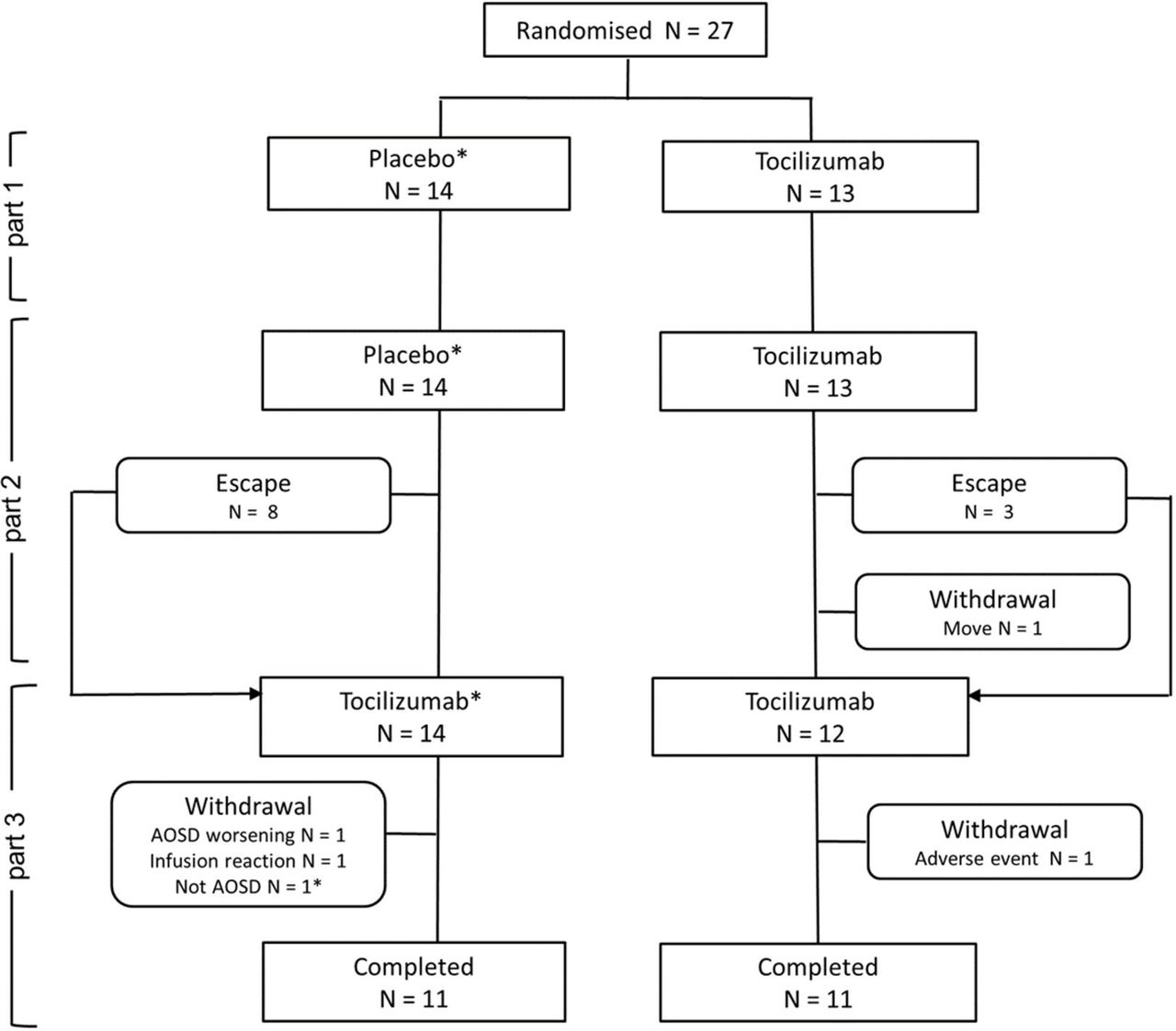
Twenty-seven patients were enrolled, of whom 14 were randomly assigned to placebo and 13 to tocilizumab. One patient who had been allocated to the placebo group was subsequently diagnosed with malignant lymphoma rather than adult-onset Still’s disease during part 3; therefore, we excluded this patient from efficacy analyses (figure 1). Eight patients who received placebo and three patients who received tocilizumab met the criteria for escape; they did not complete part 2 and moved to part 3. Five patients withdrew from the study for various reasons. Baseline demographic and disease characteristics were balanced between the groups (table 1), but patients who received placebo had slightly worse disease activity and took higher doses of glucocorticoids.

Trial profile. *The underlying disease of one participant was found to be malignant lymphoma, rather than adult-onset Still’s disease (AOSD); therefore, this participant was excluded from the efficacy analyses.
Baseline patient characteristics
Efficacy in 12-week, double-blind phase
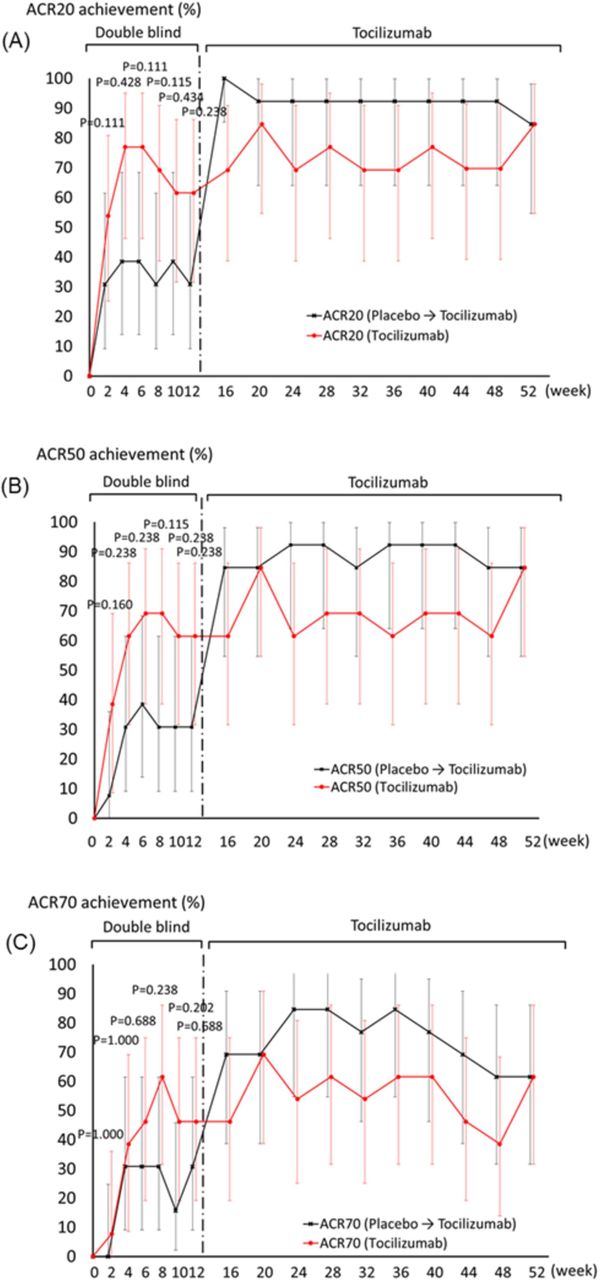
Figure 2 shows the time course transition in ACR20, ACR50 and ACR70 response rates during the study. At week 4, 61.5% (8 of 13, 95% CI 31.6 to 86.1) of patients who received tocilizumab had an ACR50 response, compared with 30.8% (4 of 13, 9.1–61.4) of patients who received placebo (p=0.238, two-tailed Fisher’s exact test). The proportion of patients with an ACR20 response was 76.9% (10 of 13) in the tocilizumab group and 38.5% (5 of 13) in the placebo group (p=0.111), whereas 38.5% (5 of 13) in the tocilizumab group and 30.8% (4 of 13) in the placebo group had an ACR70 response (p=1.000). At week 12, the proportion of patients with an ACR20 response was 61.5% (8 of 13) in the tocilizumab group, compared with 30.8% (4 of 13) in the placebo group (p=0.238). The proportion of patients with an ACR50 response was 61.5% (8 of 13) in the tocilizumab group and 30.8% (4 of 13) in the placebo group (p=0.238), whereas 46.2% (6 of 13) in the tocilizumab group and 30.8% (4 of 13) in the placebo group had an ACR70 response (p=0.688).

Time course of American College of Rheumatology (ACR) responses. Percentage of patients achieving (A) ACR20, (B) ACR50 and (C) ACR70. Bars are 95% CI.
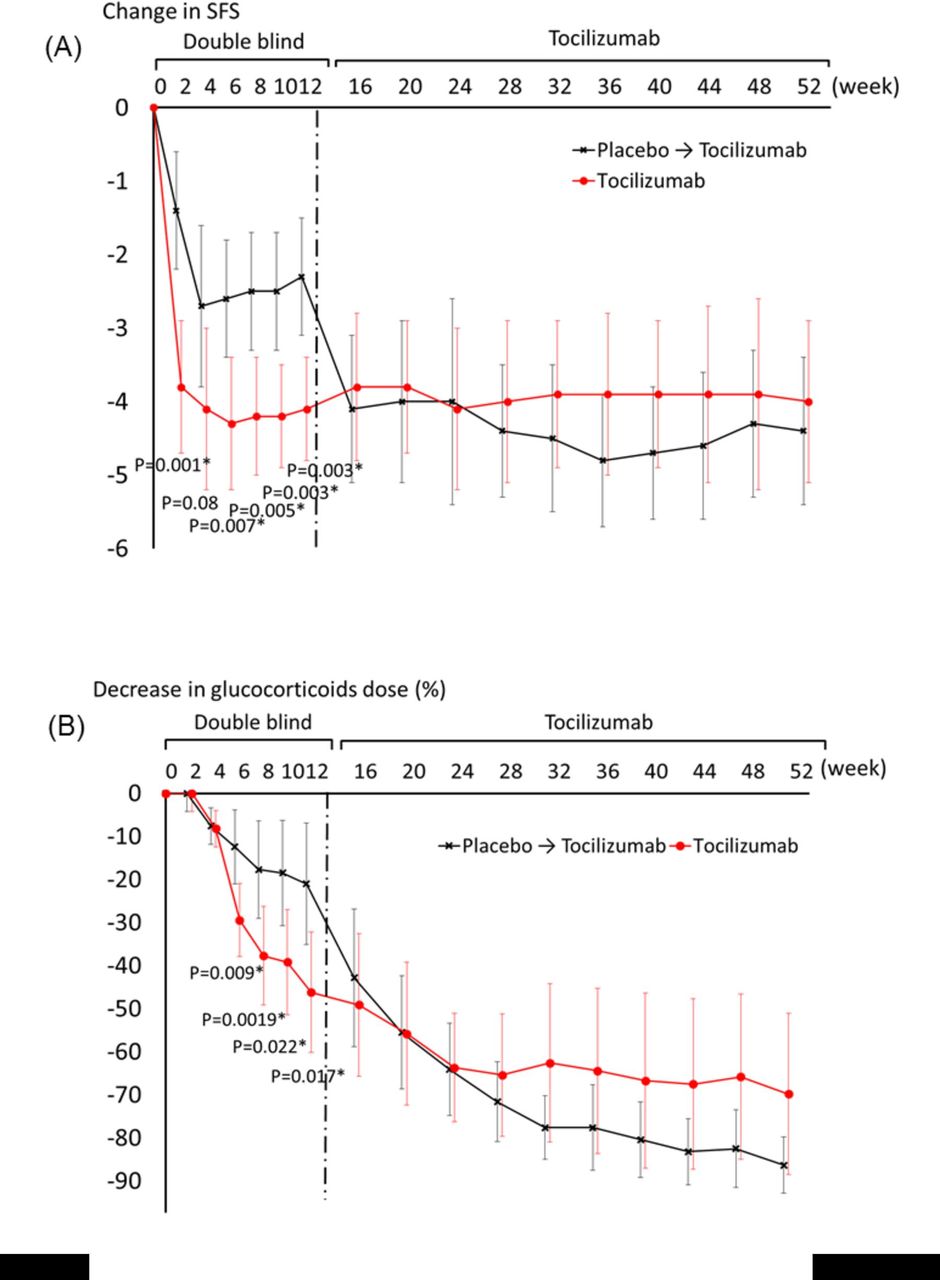
Systemic symptoms significantly improved with tocilizumab (figure 3A). The decrease in systemic feature score at week 4 from baseline in patients who received tocilizumab was –4.1, compared with –2.7 (p=0.080) in patients who received placebo. The decrease in systemic feature score at week 12 from baseline was –4.1 in the tocilizumab group and –2.3 in the placebo group (p=0.003). The results of each component of the systemic feature score are shown in online supplementary figure 1.
Supplemental material

Clinical efficacy on systemic inflammation and sparing glucocorticoid dose. (A) Changes in systemic feature score (SFS) from baseline. (B) Decrease in glucocorticoid dose from baseline. Bars are 95% CI. *P<0.05.
Tocilizumab had a significantly stronger effect on glucocorticoid sparing (figure 3B). The dose of prednisolone was decreased from baseline by 46.2% in the tocilizumab group and by 21.0% in the placebo group at week 12 (p=0.017).
For secondary endpoints at week 4, no statistical significance in between-group comparisons was observed (figure 4A–G). For secondary endpoints at week 12, significant or borderline significant differences in swollen joint count (p=0.054), Physician Visual Analogue Scale (p=0.013) and C reactive protein (p=0.030) were observed between the tocilizumab and placebo groups (figure 4A, E and G). The improvements in the other variables in the ACR core set (tender joint count, Pain Visual Analogue Scale, Patient Visual Analogue Scale and Health Assessment Questionnaire Disability Index) were not different between the groups (figure 4B, C, D and F). Serum ferritin concentrations decreased in both treatment groups (online supplementary figure 2).
Supplemental material

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Changes in other variables from baseline: (A) swollen joint count (SJC), (B) tender joint count (TJC), (C) Pain VAS, (D) Patient VAS, (E) Physician VAS, (F) Health Assessment Questionnaire Disability Index (HAQ–DI) and (G) C reactive protein (CRP). Bars are 95% CI. *P<0.05. VAS, Visual Analogue Scale.
At week 4, no patients in the tocilizumab group had fever, compared with 1 (7.7%) of 13 patients in the placebo group (p=1.000). At week 12, no patients in the tocilizumab group and 2 (15.4 %) of 13 patients in the placebo group had fever (p=0.480). Skin rash was observed in two (15.4%) patients in the tocilizumab group and five (38.5%) patients in the placebo group (p=0.378) at weeks 4 and 12. The prednisolone dose was 20.9 (±14.7) mg/day in the tocilizumab group and 29.0 (±17.4) mg/day in the placebo group at week 4, and 9.4 (±3.4) mg/day in the tocilizumab group and 16.3 (±6.8) mg/day in the placebo group at week 12.
Efficacy in 40-week, open-label phase
In part 3, the open-label phase, patients who were randomly assigned to receive tocilizumab continued to receive it and patients who were randomly assigned to receive placebo were given tocilizumab at week 12. The proportion of patients with an ACR20, ACR50 or ACR70 response (figure 2A–C) and systemic feature score in patients who switched placebo to tocilizumab improved at week 16 (figure 3A). The dose of glucocorticoids was also reduced (figure 3B) after the switch from placebo to tocilizumab. At week 52, the proportion of patients with an ACR20, ACR50 and ACR70 was 84.6% (11 of 13), 84.6% (11 of 13) and 61.5% (8 of 13), in both groups who received tocilizumab from baseline and those who received placebo and subsequently tocilizumab (figure 2A–C). The systemic feature score was 0.6±0.8 in the tocilizumab group and 0.5±0.9 in the placebo group (figure 3A). The results of each component of the systemic feature score are shown in online supplementary figure 1. The doses of prednisolone were 6.4±5.1 mg/day in the tocilizumab group with cessation in two patients and 3.3±2.4 mg/day with no cessation in the placebo group.
The change in each variable in the ACR core set became comparable between the two groups or numerically better in patients who initially received placebo owing to the baseline condition, which was slightly worse in patients receiving placebo (figure 4). The variables at week 52 were as follows: swollen joint count, 0.7±1.3 (tocilizumab) and 0.3±0.6 (placebo); tender joint count, 0.5±0.8 (tocilizumab) and 0.5±1.0 (placebo); Pain Visual Analogue Scale, 12.2±21.4 mm (tocilizumab) and 7.5±11.9 mm (placebo); Patient Visual Analogue Scale, 10.4±18.8 mm (tocilizumab) and 8.0±12.3 mm (placebo); Physician Visual Analogue Scale, 6.9±5.7 mm (tocilizumab) and 3.6±3.7 mm (placebo); Health Assessment Questionnaire Disability Index, 0.1±0.2 (tocilizumab) and 0.2±0.5 (placebo); and C reactive protein concentration, 0.0±0.0 mg/dL (tocilizumab) and 0.0±0.0 mg/dL (placebo). The time course change in ferritin concentrations is shown in online supplementary figure 2.
In part 3, six patients (three in each group) received tocilizumab at an interval of less than 10 days. The level of C reactive protein in these patients decreased from 3.3 mg/dL to 0.20 mg/dL, systemic feature score decreased from 2.3 to 0.8, and three of four patients who did not achieve ACR50 before the interval shortening fulfilled the ACR50 criteria (but were regarded as non-responders in the analysis).
Safety
In the double-blind phase, more patients in the tocilizumab group (11 patients) than in the placebo group (8 patients) had adverse events, with the most frequent adverse event being nasopharyngitis (table 2). No serious adverse events were observed. Cumulatively, 11 serious adverse events occurred in seven patients. Three serious adverse events were not related to tocilizumab: malignant lymphoma (the patient was rediagnosed with lymphoma as an underlying disease, not adult-onset Still’s disease), anaphylactic shock to contrast enhancement in CT, and drug eruption to a pain killer. Instances of serious infection included one patient with subcutaneous abscess, cellulitis and spleen abscess, one patient with cellulitis, and one patient with pneumonia. One patient was hospitalised because of adult-onset Still’s disease exacerbation, which was ameliorated by the shortening of tocilizumab interval. Aseptic necrosis in the hips of one patient was highly likely due to high-dose glucocorticoids. Anaphylactic shock to tocilizumab was observed in one patient. Infusion reaction occurred in five patients, of whom four received tocilizumab and one received placebo. Adverse events led to tocilizumab cessation in two patients. Antitocilizumab antibody was found in two patients. No death or macrophage-activated syndrome was reported. None of the patients who received tocilizumab infusion with a shorter interval than 2 weeks developed serious adverse events.
Adverse events by group
Discussion
In this double-blind, randomised, placebo-controlled phase III trial, we tried to prove the efficacy of tocilizumab in patients with adult-onset Still’s disease, but the primary endpoint of ACR50 response at week 4 did not show statistical superiority of tocilizumab to placebo because of limitations in patient accrual, study design and appropriateness of endpoints. It showed a significantly greater improvement in systemic feature score and larger decrease in glucocorticoid dose in patients who received tocilizumab than in patients who received placebo, although the validity of the statistical differences is uncertain because the study was underpowered. Despite these limitations, this has been the first randomised, placebo-controlled trial ever conducted in patients with adult-onset Still’s disease and has suggested the efficacy of anti-interleukin-6 treatment on the disease.
Although almost twice the number of patients who received tocilizumab achieved ACR50 through the double-blind phases, the primary endpoint of ACR50 response rate 4 weeks after the start of tocilizumab or placebo treatment was not statistically met. Three factors could be related to the non-significance of ACR50. One was a lack of statistical power because of the insufficient number of cases. We found it challenging to recruit patients with rare adult-onset Still’s disease who were refractory to glucocorticoids but in a non-life-threatening state who would tolerate placebo treatment, which reduced the sample size. The initial assumption of placebo achieving ACR50 of 15% was an underestimation, which also decreased the statistical power. We referred to the trial in patients with juvenile idiopathic arthritis14 to assume the 15%; however, the baseline prednisolone dose was much greater in our trial, which might partly have caused the high ACR50 achievement rate in the placebo group. Another potential reason was that tocilizumab could be more effective on systemic features than on arthritis despite our primary endpoint of ACR50, which focused on improvement in joint symptoms. Several case reports have suggested dichotomous therapeutic responses dependent on cytokines that are suppressed by biological agents, for example, articular symptoms with tumour necrosis factor antagonists, and systemic organ involvement with interleukin-1β or interleukin-6 antagonists.10 21–25 Our results are consistent with this notion, showing significant superiority to placebo on suppressing systemic inflammation. We should note, however, that although the systemic feature score we adopted in our trial significantly separated tocilizumab from placebo, assessment for systemic features in adult-onset Still’s disease has not been established yet, which is a problem to be solved in the future. One further possibility for the unmet primary endpoint is that tocilizumab dose higher than biweekly intravenous 8 mg/kg adopted in our study might be necessary. Indeed, 6 of 26 patients in our study needed tocilizumab infusion with a shorter interval to suppress inflammation. The optimal dose of tocilizumab might be different among patients, possibly depending on the soluble concentrations of interleukin-6 receptor before tocilizumab administration, as was observed in patients with rheumatoid arthritis.26
Direct inhibition of inflammatory cytokines has shown promising outcomes in the management of systemic inflammatory manifestations of adult-onset Still’s disease. Although the pathogenic mechanisms of adult-onset Still’s disease are not fully understood, the pivotal role of innate immunity involving macrophage cell activation and Th1 cytokines, such as interleukin-1β, interferon-γ, tumour necrosis factor-α, interleukin-6, interleukin-8 and interleukin-18, has been demonstrated.27–33 Most data on the benefits of suppressing cytokines have stemmed from empirical observations through small case series, with patients who were treated with currently available biological agents targeting tumour necrosis factor, interleukin-1β and interleukin-6.10 34–36 Interleukin-6 is a pleiotropic cytokine involved in both innate and acquired immunity.37 Overproduction of interleukin-6 can explain most symptoms of adult-onset Still’s disease, and high concentrations of interleukin-6 have been observed in the blood of patients with adult-onset Still’s disease. Our trial investigated tocilizumab, a monoclonal antibody targeting interleukin-6 receptor, and suggested its potential efficacy on systemic features of adult-onset Still’s disease and glucocorticoid dose reduction.
Another notable implication of this trial is that a dominant cytokine can vary according to patients. In several randomised controlled trials for systemic juvenile idiopathic arthritis conducted on tocilizumab, canakinumab, anakinra and rilonacept, some patients with systemic juvenile idiopathic arthritis responded to one drug but not to another type of drug.11–16 38 39 Moreover, there is a hypothesis that dominant cytokines may change from interleukin-1β at the beginning to interleukin-6 and subsequently to tumour necrosis factor in an individual.11 40–42 Likewise, adult-onset Still’s disease has complicated cytokine profiles. One study reported that patients with adult-onset Still’s disease could be divided into two types of disease according to distinct interleukin-6-based and interleukin-18-based cytokine profiles.32 As such, some patients with adult-onset Still’s disease might be well controlled by suppressing interleukin-18, if available. A recent, non-controlled study investigating an interleukin-18 inhibitor showed somewhat effectiveness on adult-onset Still’s disease.42
Although glucocorticoids are drugs of great importance in the treatment of adult-onset Still’s disease, its toxicity causes iatrogenic problems. The decrease in glucocorticoids dose during the disease course frequently induces flares and subsequent increases in dose, which lead to the inevitable disadvantages of glucocorticoids.2 3 In the present trial, the reduction in oral glucocorticoid dose was more than twice as much with tocilizumab as with placebo. Clinically relevant dose reductions of glucocorticoids in addition to greater decreases in systemic feature score in patients receiving tocilizumab than those receiving placebo are suggestive of the usefulness of tocilizumab in the management of adult-onset Still’s disease.
A total of 11 serious adverse events were documented during the treatment of tocilizumab. Three serious adverse events were not related to tocilizumab: malignant lymphoma (the patient was rediagnosed with lymphoma as an underlying disease, not adult-onset Still’s disease), anaphylactic shock to contrast enhancement in CT and drug eruption to a pain killer. The aseptic necrosis of the hip joints was highly likely due to high-dose glucocorticoids, and the exacerbation of adult-onset Still’s disease was ameliorated by the shortening of tocilizumab interval. Instances of serious infection, the most concerning of adverse events, included combined skin abscess, cellulitis and spleen abscess in one patient, pneumonia in one patient, and cellulitis in one patient, with the former two patients withdrawn from the trial. No death or macrophage-activated syndrome was observed. We believe that biweekly intravenous tocilizumab at 8 mg/kg in patients with adult-onset Still’s disease can be justified from the benefit–risk point of view, although careful use is essential.
Our study has some limitations. First, the sample size was small and underpowered, which led to the low statistical power resulting in the uncertainty of the results. Also, in such a study with small sample size, the imbalance in baseline characteristics might not be negligible, and the influence of one person on ratio calculations could be large, which might have caused both underestimation and overestimation. Second, the primary endpoint should have included systemic feature assessment rather than simple ACR50 to prove the efficacy of tocilizumab on systemic inflammation. Third, the double-blind period without rescue of open-label tocilizumab might be short because there was a concern about continuing placebo treatment for a longer time in patients with active adult-onset Still’s disease. Fourth, although we did not set the upper limit of disease activity, the exclusion criteria regarding thrombocytopaenia and transaminase elevation might have excluded eligible but severely ill patients. In addition, investigators must have selected patients at enrolment who they thought would tolerate placebo treatment. These could have led to selection bias. Fifth, the high response to placebo might be due to the short duration of prior glucocorticoid treatment and the baseline glucocorticoid dose. Sixth, the optimal usage of glucocorticoids at tocilizumab initiation is unclear. Patients in the placebo group received higher doses of baseline glucocorticoids and a longer period of glucocorticoid treatment without dose reduction at tocilizumab initiation, and in this context it appears that higher ACR responses were obtained in the placebo group than in the tocilizumab group in the open-label phase. Despite these limitations, this is the first double-blind, randomised, placebo-controlled trial demonstrating the efficacy of tocilizumab on relieving systemic inflammation and glucocorticoid sparing, and our findings provide useful insights into the management of adult-onset Still’s disease.
In conclusion, we suggest that inhibiting interleukin-6 with tocilizumab is effective in patients with adult-onset Still’s disease refractory to glucocorticoid therapy on suppressing systemic features and glucocorticoid dose reduction. However, the efficacy was not proven in our trial because of limitations in patient accrual, study design and appropriateness of endpoints. A well-designed, properly powered study will substantiate our preliminary findings.
Acknowledgments
The authors acknowledge and thank all investigators, participants and staff in this study. Shinsuke Yasuda (Hokkaido University, Sapporo, Japan) and Takayuki Sumida (Tsukuba University, Tsukuba, Japan) are other investigators of this trial.
References
Footnotes
Handling editor Josef S Smolen
Contributors YK, HK, TA and TT designed the study in collaboration with the Japanese Pharmaceuticals and Medical Devices Agency. YK, KI, TI, KM, HT, YT and TT were involved in patient recruitment, collecting data and managing their clinical research sites. All authors were involved in writing the manuscript and approved the final version. The corresponding author had full access to all the data in the study and had final responsibility for the decision to submit for publication.
Funding This study was supported by the Clinical Trial on Development of New Drugs and Medical Devices from the Japanese Ministry of Health, Labour and Welfare under grant number H23-実用化(臨床)-指定-005, and the Japan Agency for Medical Research and Development under grant number JP15lk0103004. Treatment drugs were provided by Chugai Pharmaceutical. The funders had no role in the study design, data collection, data analysis, data interpretation or writing of the report.
Competing interests YK has received grants or speaking fees from AbbVie, Astellas, Ayumi, Bristol-Myers Squibb, Chugai, Eisai, Eli Lilly, Hisamitsu, Janssen, Kissei, Pfizer, Sanofi, Takeda, Mitsubishi-Tanabe and UCB. HK has received grants or speaking fees from AbbVie, Ayumi, Chugai, Mitsubishi-Tanabe, Astellas, Takeda, Eisai, Novartis, Sanofi, Eli Lilly, GlaxoSmithKline, Bristol-Myers Squibb, Pfizer and Janssen. KI has received grants or speaking fees from Tanabe-Mitsubishi Pharma, AbbVie, Bristol-Myers KK, Eisai, UCB, Pfizer Japan, Takeda Pharmaceutical, Eli Lilly, Astellas Pharma, Chugai Pharmaceutical and Kyowa Hakko Kirin. IT has received grants or speaking fees from Chugai, Ono, Pfizer, Mitsubishi-Tanabe and Astellas. KM has received grants or speaking fees from AbbVie, Chugai, Mitsubishi-Tanabe, Astellas, Takeda, Eisai, UCB, Daiichi Sankyo, Eli Lilly, GlaxoSmithKline, Bristol-Myers Squibb, Pfizer and Janssen. HT has received grants or speaking fees from Pfizer, Daiichi Sankyo and Mitsubishi-Tanabe. YT has received grants or speaking fees from Mitsubishi-Tanabe, Takeda, Bristol-Myers, Chugai Pharmaceutical, Astellas Pharma, AbbVie, MSD, Eli Lilly, YL Biologics, Daiichi Sankyo, Sanofi, Janssen, Pfizer, Kyowa Kirin, Eisai and Ono. HT and TA have nothing to declare. TT has received research grants or speaking fees from Astellas Pharma, Bristol-Myers KK, Chugai Pharmaceutical, Daiichi Sankyo, Takeda Pharmaceutical, Teijin Pharma, AbbVie GK, Asahi Kasei Pharma, Mitsubishi-Tanabe Pharma, AstraZeneca KK, Eli Lilly Japan KK, Novartis Pharma KK, AbbVie GK, Nippon Kayaku, Janssen Pharmaceutical KK, Taiho Pharmaceutical and Pfizer Japan.
Patient consent Obtained.
Ethics approval The institutional review board at each site approved the study.
Provenance and peer review Not commissioned; externally peer reviewed.