Article Text

Abstract
Background Nintedanib is an inhibitor targeting platelet-derived growth factor receptor, fibroblast growth factor receptor and vascular endothelial growth factor receptor tyrosine kinases that has recently been approved for the treatment of idiopathic pulmonary fibrosis. The aim of this study was to analyse the effects of nintedanib in the fos-related antigen-2 (Fra2) mouse model of systemic sclerosis (SSc).
Methods The effects of nintedanib on pulmonary arterial hypertension with proliferation of pulmonary vascular smooth muscle cells (PVSMCs) and luminal occlusion, on microvascular disease with apoptosis of microvascular endothelial cells (MVECs) and on fibroblast activation with myofibroblast differentiation and accumulation of extracellular matrix were analysed. We also studied the effects of nintedanib on the levels of key mediators involved in the pathogenesis of SSc and on macrophage polarisation.
Results Nintedanib inhibited proliferation of PVSMCs and prevented thickening of the vessel walls and luminal occlusion of pulmonary arteries. Treatment with nintedanib also inhibited apoptosis of MVECs and blunted the capillary rarefaction in Fra2-transgenic mice. These effects were associated with a normalisation of the serum levels of vascular endothelial growth factor in Fra2 mice on treatment with nintedanib. Nintedanib also effectively blocked myofibroblast differentiation and reduced pulmonary, dermal and myocardial fibrosis in Fra2-transgenic mice. The antifibrotic effects of nintedanib were associated with impaired M2 polarisation of monocytes and reduced numbers of M2 macrophages.
Conclusion Nintedanib targets core features of SSc in Fra2-transgenic mice and ameliorates histological features of pulmonary arterial hypertension, destructive microangiopathy and pulmonary and dermal fibrosis. These data might have direct implications for the ongoing phase III clinical trial with nintedanib in SSc-associated interstitial lung disease.
- scleroderma
- endothelial
- macrophages
- VEGF
- CSF
Statistics from Altmetric.com
Systemic sclerosis (SSc) is characterised by vascular remodelling with loss of capillary and pulmonary arterial hypertension (PAH) and progressive tissue fibrosis of the skin and internal organs such as the lungs.1 Although therapies are approved for the treatment of vascular manifestations, targeted therapies for the treatment of fibrosis in SSc are not yet available for clinical use.2
Nintedanib, which has recently been approved for the treatment of idiopathic pulmonary fibrosis (IPF), may be an interesting candidate for antifibrotic therapies in SSc. Nintedanib is a potent inhibitor of platelet derived growth factor receptor (PDGFR)-α and PDGFR-β, vascular endothelial growth factor receptor (VEGFR)-1, 2, 3, fibroblast growth factor receptor (FGFR)-1, 2, 3 and SRC family kinases.3 Recent data demonstrate that nintedanib in pharmacologically relevant concentrations effectively inhibits colony-stimulating factor (CSF)-receptor.4 All of those pathways have been implicated into the pathogenesis of fibrosis and have been discussed as candidates for targeted therapies in SSc.5–8 Nintedanib may thus offer the potential to simultaneously inhibit multiple profibrotic pathways with a single drug. Indeed, we demonstrated potent antifibrotic effects of nintedanib in several preclinical models of skin fibrosis9 and a phase III clinical trial with nintedanib in SSc-associated interstitial lung disease is currently ongoing (SENSCIS trial; NCT02597933). However, the effects of nintedanib on vascular manifestations have not been investigated so far.
While its inhibitory effects on PDGFRs may be beneficial in PAH, the effects of nintedanib on microvascular disease in SSc are of concern. Given that PDGF, VEGF and FGF signalling as well as SRC kinases have all been implicated in angiogenesis,10 11 nintedanib may interfere with vascular repair and regeneration. Although no adverse events related to inhibition of angiogenesis have been observed in clinical trials with patients with IPF,12 patients with SSc might be more sensitive due to the pre-existing microvascular disease. On the other hand, accumulating evidence suggests that uncontrolled VEGF signalling in SSc may actually promote microvascular disease and capillary loss in SSc.5 7
Fos-related antigen-2 transgenic (Fra2 transgenic) mice resemble the core clinical features of SSc as they display fibrotic and vascular manifestations of SSc.13–16 Fra2 transgenic mice develop a destructive microvascular disease with apoptosis of endothelial cells followed by systemic fibrotic manifestations and PAH. In this study, we used Fra2 transgenic mice as a model system to study the effects of nintedanib on vascular and fibrotic manifestations of SSc.
Materials and methods
Fra2 transgenic mice
Treatment of Fra2 transgenic mice with nintedanib was initiated at an age of 10 weeks and mice were sacrificed at an age of 16 weeks. Non-transgenic littermates served as controls. Each group consisted of six mice. Nintedanib was administered by oral gavage. Controls received vehicle treatment.
Quantification of pulmonary, dermal and cardiac fibrosis
Skin fibrosis was assessed histologically and in addition by quantification of the hydroxyproline content and analyses of myofibroblast counts. Interstitial lung changes were analysed histologically by H&E and trichrome staining, by quantification of myofibroblasts, by immunofluorescence and biochemically by determination of hydroxyproline contents.15 17–20 Assessment of cardiac fibrosis also followed established protocols.21
Evaluation of vascular remodelling of the pulmonary arteries
Vascular changes were evaluated according to the Dana Point consensus criteria using established protocols and readouts.22 23 Muscular blood vessels were identified by positive staining for Smooth Muscle Protein 22-alpha, green (α-SMA) and α smooth muscle actin (SM22α). While α-SMA is also expressed by myofibroblasts, the expression of SM22α is restricted to vascular smooth muscle cells. The vessel wall thickness of pulmonary arteries was measured on H&E stained sections captured at 400-fold magnification. For analysis, only the circular-shaped vessels were included, the oblique and longitudinal sectioned vessels were excluded. The thickness of vessel walls was evaluated by a minimum of three measurements per vessel14 by using the following equation: (outside diameter-inside diameter)/outside diameter. The results were express as x-fold changes.24 As an additional outcome for vascular remodelling, the degree of luminal occlusion of pulmonary arteries (α-SMA and SM22α-positive) was examined by counting the numbers of occluded lumina/high-power field (HPF).25 The percentage of proliferating vascular smooth muscle cells was evaluated by triple staining for 4′,6-Diamidin-2-phenylindol (DAPI) (visualisation of nuclei), Ki67 (proliferation marker) and SM22α.13 14 16
Immunofluorescence stainings
M2 macrophages can be identified by immunohistochemistry using a combination of pan-macrophage markers (F4/80 in mice) and prototypical M2 markers such as arginase and cMAF, a transcription factor that has recently been shown to be required for the expression of arginase.28 M1 macrophages were defined by staining for inducibile nitric oxide synthase (iNOS), CD11c and F4/80. According to established protocols,29 the following primary antibodies were used: F4/80 (Biorad, Puchheim, Germany), arginase (Santa Cruz, San Diego, California, USA), cMAF (Abgent, San Diego, California, USA) iNOS (Invitrogen) and CD11c (Abcam, Cambridge, UK). In a subset of experiments, costainings of F4/80 with either interleukin (IL)-12 (LSBIO, Seattle, Washington, USA), IL-4 (Santa Cruz) or IL-13 (Bioss, Woburn, Massachusetts, USA) were performed. Additional stainings included vimentin (mesenchymal cells, Merck, Darmstadt, Germany) and CD45 (leukocytes (Abcam)). The following secondary antibodies were used: Alexa Fluor 647 chicken anti-rat (Invitrogen, Germany), Alexa Fluor 488 donkey anti-rabbit (Abcam), Alexa Fluor 594 donkey anti-goat (Invitrogen).20 Sections were counterstained with DAPI.
Quantification of the staining intensity
The staining intensity was quantified as described using ImageJ.14 A density threshold was set to quantify the positive staining by using the respective negative controls. The threshold was selected to exclude unspecific background staining. The same thresholds and system settings were used for all slides. The number of pixels falling within the threshold, indicating the quantity of staining positivity, was recorded for each field.
Isolation, selection and culture of macrophages
Peripheral blood mononuclear cells (PBMCs) were isolated from six healthy volunteers using Lymphoflot (Bio-Rad, Hercules, California, USA). CD14-microbeads (Miltenyi Biotech, Bergisch-Gladbach, Germany) were used for positive selection of human monocytes and macrophages.26 Monocytes/macrophages were seeded in RPMI medium supplemented with 10% fetal bovine serum (FBS) (all Gibco, Basel, Switzerland) with or without 25 nM human M-CSF (Peprotech, Rocky Hill, New Jersey, USA) for 6 days at 37°C and 5% CO2. At days 3 and 6, the medium was replaced by fresh medium. At day 6, cells were treated with 100 nM of nintedanib. Twenty-four hours later, cells were stimulated with 10 nM IL−4 and 10 nM IL-13 (both Peprotech). Twenty-four hours after stimulation, the expression of M1 and M2 markers was measured.
Microtitre tetrazolium assay
The microtitre tetrazolium [3, (4, 5-dimethylthiazol-2-yl)2, 5-diphenyl-tetrazolium-bromide] assay is an established method to analyse proliferation. The effect of nintedanib on primary human vascular smooth muscle cells was analysed in the presence or absence of PDGF.
Cytokine measurements
Cytokines in the serum of mice were quantified using the mouse inflammation multianalyte profile, which is based on ultrasensitive immunoassays for 37 cytokines (Myriad-RBM, Austin, Texas, USA).
Statistics
All data are presented as median±IQR. Differences were analysed using the Mann-Whitney U test. p Values are expressed as follows: 0.05 > p≥0.01 as *; 0.01 > p≥0.001 as **; p<0.001 as ***.
Results
Treatment with nintedanib reduces dermal, pulmonary and myocardial fibrosis in Fra2 transgenic mice
We first evaluated the effect of nintedanib on fibrosis of the skin and the lungs. Treatment with nintedanib was well tolerated and reduced the weight loss in Fra2 transgenic mice (see online supplementary figure S1). We did not observe changes in activity, in texture of the fur or in the consistency of the stool at both doses.
Supplementary file 1
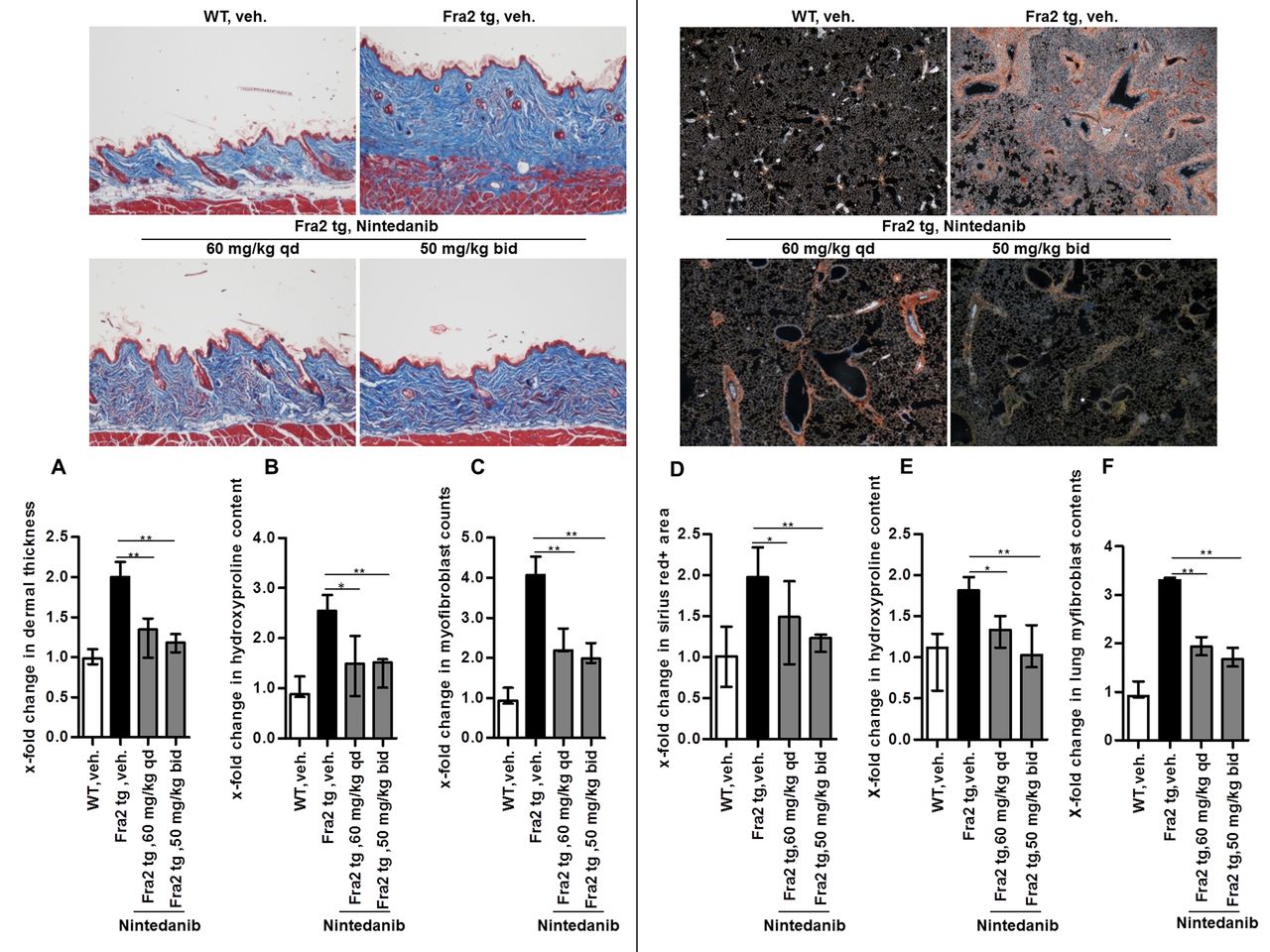
Doses of 60 mg/kg qd or 50 mg/kg/two times per day strongly ameliorated skin fibrosis. Both doses of nintedanib were equally effective and significantly reduced dermal thickening, collagen accumulation and myofibroblast differentiation as compared with vehicle-treated Fra2 transgenic mice (figure 1A–C).

Nintedanib reduces dermal and pulmonary fibrosis in fos-related antigen-2 (Fra2) transgenic mice. (A–C) Effects on skin fibrosis. Representative images of Masson trichrome-stained skin sections at 100-fold magnification and quantification of dermal thickness (A), hydroxyproline content (B) and myofibroblast count (C). (D–F) Effects on pulmonary fibrosis. Representative images of Sirius red-stained sections of lungs at 40-fold magnification and quantification of the fibrotic area (D), of the hydroxyproline content (E) and of myofibroblast counts (F). *0.01≤p<0.05; **0.001≤p<0.01 versus vehicle-treated Fra2-transgenic mice (Fra2 tg). WT, vehicle-treated wild-type mice, n=6 mice per group.
Nintedanib-treated mice also demonstrated reduced pulmonary fibrosis with decreased fibrotic area, lower hydroxyproline content and decreased myofibroblast counts compared with control mice (figure 1D–F).
Myocardial changes in patients with SSc are mimicked in Fra2 transgenic mice.21 Treatment with nintedanib at 60 mg/kg/qd reduced the extent of fibrosis, ameliorated perivascular inflammation and decreased apoptosis of endothelial cells (see online supplementary figure S2A–F).
Supplementary file 2
Nintedanib inhibits remodelling of the pulmonary arteries
Fra2 transgenic mice also develop pulmonary arterial hypertension with extensive remodelling of the pulmonary arteries. Both doses of nintedanib significantly reduced thickening of the walls of pulmonary arteries (figure 2A, B) and decreased the number of occluded vessels compared with vehicle-treated Fra2 transgenic mice (figure 2C). Consistent with these findings, the number of proliferating vascular smooth muscle cells was strongly decreased in nintedanib-treated mice (figure 2D, E). We observed a trend towards higher efficacy with nintedanib at 50 mg/kg/two times per day as compared with 60 mg/kg/qd, which, however, did not reach statistical significance. Nintedanib in a concentration-dependent manner also inhibited proliferation of human pulmonary vascular smooth muscle cells in vitro, both under basal conditions and on stimulation with PDGF (figure 2F).

Treatment with nintedanib reduces vascular remodelling of the pulmonary arteries in Fra2 transgenic mice. (A) Representative images of H&E-stained lung sections with thickened vessel walls shown at 400-fold magnification. Arrows indicate thickened vessel walls, stars indicate occluded vessels. (B) Percentage of occluded vessels. (C) Average thickness of the vessel wall. (D) Representative examples of triple staining for DAPI (nuclear staining), Ki67 (proliferation marker) and SM22α (vascular smooth muscle cells) at 200-fold and 600-fold magnification. (E) Relative proportion of proliferating vascular smooth muscle cells per total number of vascular smooth muscle cells. (F) Effects of nintedanib on the proliferation of human pulmonary artery vascular smooth muscle cells at basal conditions and on stimulation with platelet-derived growth factor (PDGF). *0.01≤p<0.05; **0.001≤p<0.01; ***p<0.001 versus vehicle-treated Fra2 transgenic mice. DAPI, 4′,6-diamidin-2-phenylindol, blue; Fra2 tg, Fra2-transgenic mice; SM22α, smooth muscle protein 22-alpha, green; WT, vehicle-treated wild-type mice.
Nintedanib ameliorates microvascular disease in Fra2 transgenic mice
Another characteristic feature of Fra2 transgenic mice is apoptosis of endothelial cells with subsequent loss of capillaries. The number of apoptotic endothelial cells in the skin and lungs of Fra2 transgenic mice was reduced by treatment with nintedanib (figure 3A, B and online supplementary figure S3). Consistently, capillary loss was reduced and increased numbers of vessels in the skin were observed in the dermis of nintedanib-treated mice (figure 3C). As for remodelling of pulmonary arteries, doses of 50 mg/kg/two times per day tended to be more effective than 60 mg/kg/qd and statistically significant differences were found for total microvessel counts between both groups.
Supplementary file 3

Nintedanib ameliorates microvascular manifestations in fos-related antigen-2 (Fra2) transgenic mice. (A) Representative images of skin sections stained for TdT-mediated dUTP-biotin nick end labeling (TUNEL) (apoptosis marker), CD31 (endothelial cells) and 4′,6-diamidin-2-phenylindol (DAPI) (nuclear staining) shown at 400-fold magnification. (B) Quantification of apoptotic microvascular endothelial cells in the skin. (C) Relative numbers of CD31-positive vessels. n=6 mice per group. *0.01≤p<0.05 versus vehicle-treated Fra2 transgenic mice. Fra2 tg, Fra2-transgenic mice; WT, vehicle-treated wild-type mice.
Treatment with Nintedanib normalises the levels of M-CSF1 and VEGF in Fra2 transgenic mice
To gain additional insights into the mechanisms underlying the antifibrotic and vasoprotective effects of nintedanib in the Fra2 model, we screened for differences in the serum levels of central inflammatory, fibrotic and angiogenic mediators. Several of these cytokines and growth factors such as epithelial growth factor (EGF), IL-1β, IL-5, IL-6, IL-10, IL-18, IP-10, MCP-1, MCP-3, MCP-5, M-CSF, macrophage inflammatory protein (MIP)-1α, MIP-1β, stem cell factor (SCF), tissue inhibitor of MMP (TIMP)-1, thrombopoietin and VEGF were upregulated in Fra2 transgenic mice as compared with non-transgenic littermates (figure 4A). Of those, two mediators were significantly affected by treatment with nintedanib. Treatment with nintedanib decreased the levels of M-CSF (figure 4B) and of VEGF (at a dose of 60 mg/kg/qd) in Fra2 transgenic mice (figure 4C).

Treatment with nintedanib alters the levels of macrophage (M)-colony-stimulating factor (CSF) and vascular endothelial growth factor (VEGF) and reduces M2 macrophage counts in fos-related antigen-2 (Fra2) transgenic mice. (A) Serum levels of major inflammatory, fibrotic and angiogenic mediators in Fra2 transgenic mice and littermate controls in pg/mL. Effects of nintedanib on the levels of M-CSF1 (B) and VEGF (C) in Fra2 transgenic mice. n=6 mice per group. (D) Fold changes in M2 macrophage counts defined as F4/80, cMAF, arginase triple-positive cells. (E) Fold changes in M1 macrophage counts defined as F4/80, iNOS, CD11c triple-positive cells. *0.01≤p<0.05; **0.001≤p<0.01 versus vehicle-treated Fra2 transgenic mice. Fra2 tg, Fra2-transgenic mice; HPF, high-power field; IL, interleukin; WT, vehicle-treated wild-type mice.
Nintedanib inhibits M2 differentiation of monocytes
M-CSF is a central growth factor for monocytes and macrophages and favours their alternative activation and differentiation into M2 macrophages.30 M2 macrophages are implicated into the pathogenesis of fibrosis.31 32 Changes in the expression of M2 genes have recently been linked to clinical responses in tocilizumab-treated patients with SSc.33 34 Based on the downregulation of M-CSF, we hypothesised that nintedanib may inhibit M2 polarisation in Fra2 transgenic mice. Indeed, we observed highly increased numbers of M2 macrophages in the skin of Fra2 transgenic mice compared with wildtype (WT) littermates. Treatment with nintedanib completely prevented the increase in M2 macrophages with M2 counts comparable to those in non-transgenic control (figure 4D and online supplementary figure S4). In contrast to M2 macrophages, the number of M1 macrophages (defined by the expression of iNOS and CD11c in combination with F4/80) did not differ between Fra2 transgenic mice and non-transgenic littermates and M1 macrophage counts were not affected by treatment with nintedanib (figure 4E).
Supplementary file 4
To confirm that inhibitory effects of nintedanib are not restricted to the Fra2 model or to murine monocytes, we tested the effects of nintedanib on alternative activation of human monocytes induced by M-CSF1, IL-4 and IL-13. Nintedanib reduced M2 counts with suppression of the expression levels of individual M2 markers such as CD163 or CD206 (figure 5A). The expression of M1 markers such as CD86, toll-like receptor 4 (TLR4) and human leukocyte antigen-DR (HLA-DR) (figure 5B) or general markers of monocytes such as CD14 was unchanged or even increased by incubation with nintedanib (data not shown). The effects of nintedanib on macrophage polarisation could be explained by its direct inhibitory effects on CSF1R (recent, unpublished information by Boehringer Ingelheim). In support of this hypothesis, individual inhibition of PDGFR, FGFR or VEGFR did not affect M2 polarisation of macrophages (see online supplementary file S5A and B). Together, these data demonstrate that nintedanib inhibits M2 polarisation of macrophages, likely by direct inhibition of CSF1R.
Supplementary file 5

![[SP1.jpg]](https://ard.bmj.com/content/annrheumdis/76/11/1941/DC1/embed/inline-supplementary-material-1.jpg?download=true){kind=link}
{kind=link}
![[SP2.jpg]](https://ard.bmj.com/content/annrheumdis/76/11/1941/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
{kind=link}
![[SP3.jpg]](https://ard.bmj.com/content/annrheumdis/76/11/1941/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
{kind=link}
{kind=link}
![[SP4.jpg]](https://ard.bmj.com/content/annrheumdis/76/11/1941/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
![[SP5.jpg]](https://ard.bmj.com/content/annrheumdis/76/11/1941/DC5/embed/inline-supplementary-material-5.jpg?download=true){kind=link}
{kind=link}
Nintedanib inhibits alternative activation of monocytes. (A) Effects of nintedanib on M2 polarisation of human macrophages incubated with macrophage (M)-colony-stimulating factor (CSF), interleukin (IL)-4 and IL-13 as analysed by changes in the mean fluorescence intensity of CD163and CD206. (B) Expression levels of M1 markers CD86,toll-like receptor 4 (TLR4) and human leukocyte antigen-DR (HLA-DR). (C) Fold changes in the number of IL-4, IL-13 and IL-12 positive macrophages in fos-related antigen-2 (Fra2) transgenic mice with or without nintedanib treatment. *0.01≤p<0.05; **0.001≤p<0.01.
To confirm the functional relevance of those findings, we analysed the release of M1 and M2 cytokines in Fra2 transgenic mice. The numbers of macrophages positive for IL-4 or for IL-13 were strongly increased in Fra2 transgenic mice. Treatment with nintedanib reduced the number of IL-4 and IL13-positive macrophages (figure 5C) back to the levels of non-transgenic mice. In contrast, the number of IL-12 positive macrophages was not increased in Fra2-transgenic mice and was not affected by treatment with nintedanib (figure 5C). Similar results were obtained for the total numbers of IL-4, IL-13 and IL-12 positive cells (data not shown).
Discussion
Fra2 transgenic mice resemble the clinical manifestations of SSc and develop not only systemic fibrotic disease, but also SSc-like microvascular disease and PAH.13–16 Evidence provided by studies on the efficacy of imatinib, which rather selectively targets PDGF signalling and cellular abelson kinase (c-ABL) in fibrotic diseases, indicates that Fra2 transgenic mice may reflect the activation levels of profibrotic pathways in human SSc more closely and may better predict responses as compared with other mouse models.23 35–37 Indeed, the tyrosine kinase inhibitor imatinib that did not reach its primary end point in clinical trials in SSc was also not effective in Fra2 transgenic mice, but reduced fibrosis induced by bleomycin.23 We now demonstrate that nintedanib, which inhibits FGFRs, PDGFRs, VEGFRs, SRC family kinases and, according to very recent data, also CSF1R, effectively reduces dermal and pulmonary fibrosis in Fra2 transgenic mice, thereby extending previous results on cultured fibroblasts and localised skin fibrosis.9
In addition to its direct effects on fibroblasts, we provide here novel evidence that nintedanib may also target fibroblast activation indirectly by blocking alternative activation and M2 polarisation of macrophages. Nintedanib inhibits M2 polarisation of healthy human macrophages in vitro and also strongly reduces M2 macrophage counts in Fra2 transgenic mice. These inhibitory effects of nintedanib on macrophage polarisation are likely mediated by direct inhibition of CSF1R. Selective inhibition of PDGFR, VEGFR or FGFR did not block M2 polarisation in vitro, whereas targeted inhibition of CSF1R is known to interfere with the alternative activation of macrophages.38 However, confirmation of the findings with monocytes from patients with SSc is warranted. Given that M2 macrophages are a rich source of profibrotic mediators34 and M2 macrophages have been shown to strongly affect the outcome in various preclinical models of fibrosis39 and that changes in M2 mRNA signature have recently been linked to clinical outcomes in patients with SSc,20 40 the effects of nintedanib on M2 polarisation might significantly contribute to the antifibrotic effects of nintedanib in vivo and may be of direct clinical relevance.
Although our study is limited by the lack of functional assessment by echocardiography or right heart catheter, we provide clear evidence that treatment with nintedanib also inhibited the proliferation of pulmonary vascular smooth muscle cells and ameliorated the histological features of PAH. Further studies are required to unravel whether those effects are solely mediated by inhibition of PDGFR or whether other targets of nintedanib contribute to the potent inhibitory effects. Despite the technical challenges of right heart catheterisation in mice, i.p. with widespread organ involvement as in Fra2 transgenic mice, right heart catheterisation in follow-up studies would further confirm the beneficial effects of nintedanib on PAH. At the molecular level, inhibition of PDGFR with impaired proliferation of vascular smooth muscle cells is likely to play a central role for the beneficial effects of nintedanib on PAH. As recent data provide elegant evidence that apoptosis of endothelial cell may promote PAH,39 further studies should investigate whether the positive effects of nintedanib on the histological features of PAH are in part mediated by inhibition of pulmonary vascular EC apoptosis.
Treatment with nintedanib also reduced apoptosis of microvascular endothelial cells of the skin and ameliorated the loss of capillaries in Fra2 transgenic mice. These findings seem unexpected on first view given the inhibition of multiple proangiogenic mediators by nintedanib. However, excessive upregulation of angiogenic factors such as VEGF has previously been shown to perturb, rather than to promote angiogenesis and to induce microvascular alterations reminiscent of those observed in SSc.5 7 Treatment with nintedanib may thus improve microvascular manifestations in Fra2 transgenic mice by preventing the deleterious effects of excessive, uncontrolled activity of angiogenic factors. This hypothesis is supported by the finding that the levels of VEGF are upregulated in Fra2 transgenic mice and that treatment with nintedanib in doses of 60 mg/kg/qd normalised the levels of VEGF. Mechanistically, nintedanib may inhibit VEGF expression by inhibiting inflammation and in particular macrophage activation, as macrophages are a major source of VEGF.41 42 The effects of nintedanib on M-CSF may have also contributed to the beneficial effects on vascular alterations. M-CSF has been shown to be capable of modulating angiogenesis directly, but can also regulate the activation of endothelial cells indirectly by inducing the expression of VEGF.43 However, further studies are required to decipher the molecular regulation of VEGF by nintedanib. Moreover, it will be crucial to assess the effects of nintedanib on angiogenesis during wound healing that demands increased formation of new vessels to initiate tissue repair.
Apart from potential effects on physiological wound healing, common side effects of nintedanib with particular relevance to SSc are gastrointestinal adverse events. Although mild in most patients, gastrointestinal side effects were common in IPF studies and may complicate gastrointestinal involvement in SSc.
In summary, our data provide preclinical evidence that treatment with nintedanib may not only improve fibrosis of skin, lungs and heart, but also ameliorate vascular manifestations as the other major cause of morbidity and mortality in SSc. We also provide evidence that nintedanib may not only exert its antifibrotic effects by direct inhibition of fibroblast activation, but also by inhibition of alternative activation of macrophages. It will be important to follow-up on these preclinical findings in the ongoing clinical study and to carefully monitor microvascular disease and PAH.
Acknowledgments
We thank Regina Kleinlein and Katja Dreißigacker for excellent technical assistance.
References
Footnotes
Handling editor Tore K Kvien
Contributors Design of the study: LW, JHWD. Acquisition of data: JH, CM, YZ, AS, CD, CB, CWC. Interpretation of data: JH, CM, YZ, AS, CD, CB, CWC, UH, OD, GS, LW, JHWD. Manuscript preparation: JH, CM, LW, JHWD.
Funding Grants DI 1537/5-1, DI 1537/7-1, DI 1537/8-1, DI 1537/9-1, DI 1537/11-1, DE 2414/2-1 andAK 144/2-1 of the Deutsche Forschungsgemeinschaft, grants A57, and A64 of the IZKF inErlangen, grant 2013.056.1 of the Wilhelm-Sander-Foundation, grants 2014_A47 and2014_A248 of the Else-Kröner-Fresenius-Foundation, grant 2013.056.1 of the Wilhelm SanderFoundation and a Career Support Award of Medicine of the Ernst Jung Foundation.
Competing interests OD has consultancy relationships and/or has received research funding from Actelion, Pfizer, Ergonex, BMS, Sanofi-Aventis, United BioSource Corporation, Roche/Genentech, Medac, Biovitrium, Boehringer Ingelheim, Novartis, 4D Science, Active Biotech, Bayer, Sinoxa, Serodapharm, EpiPharm, GSK, Pharmacyclics and Biogen. LW is an employee of Boehringer-Ingelheim. JHWD has consultancy relationships with Actelion, Active Biotech, Anamar, Bayer Pharma, Boehringer Ingelheim, Celgene, Galapagos, GSK, Inventiva, JB Therapeutics, Medac, Pfizer, RuiYi and UCB. JHWD has received research funding from Anamar, Active Biotech, Array Biopharma, BMS, Bayer Pharma, Boehringer Ingelheim, Celgene, GSK, Novartis, Sanofi-Aventis and UCB. JHWD is stock owner of 4D Science.
Patient consent Obtained.
Ethics approval Ethical Committee of the University of Erlangen-Nuremberg.
Provenance and peer review Not commissioned; externally peer reviewed.
Correction notice This article has been corrected since it published Online First. The title has been corrected to: Nintedanib inhibits macrophage activation and ameliorates vascular and fibrotic manifestations in the Fra2 mouse model of systemic sclerosis.