Article Text

Abstract
Objectives An imbalance between neutrophil extracellular trap (NET) formation and degradation has been described in systemic lupus erythematosus (SLE), potentially contributing to autoantigen externalisation, type I interferon synthesis and endothelial damage. We have demonstrated that peptidylarginine deiminase (PAD) inhibition reduces NET formation and protects against lupus-related vascular damage in the New Zealand Mixed model of lupus. However, another strategy for inhibiting NETs—knockout of NOX2—accelerates lupus in a different murine model, MRL/lpr. Here, we test the effects of PAD inhibition on MRL/lpr mice in order to clarify whether some NET inhibitory pathways may be consistently therapeutic across models of SLE.
Methods NET formation and autoantibodies to NETs were characterised in lupus-prone MRL/lpr mice. MRL/lpr mice were also treated with two different PAD inhibitors, Cl-amidine and the newly described BB-Cl-amidine. NET formation, endothelial function, interferon signature, nephritis and skin disease were examined in treated mice.
Results Neutrophils from MRL/lpr mice demonstrate accelerated NET formation compared with controls. MRL/lpr mice also form autoantibodies to NETs and have evidence of endothelial dysfunction. PAD inhibition markedly improves endothelial function, while downregulating the expression of type I interferon-regulated genes. PAD inhibition also reduces proteinuria and immune complex deposition in the kidneys, while protecting against skin disease.
Conclusions PAD inhibition reduces NET formation, while protecting against lupus-related damage to the vasculature, kidneys and skin in various lupus models. The strategy by which NETs are inhibited will have to be carefully considered if human studies are to be undertaken.
- Systemic Lupus Erythematosus
- Autoimmune Diseases
- Autoimmunity
Statistics from Altmetric.com
Introduction
Patients with systemic lupus erythematosus (SLE) form autoantibodies to nuclear antigens. The resulting immune complexes (ICs) stimulate type I interferon (IFN) production by plasmacytoid dendritic cells (pDCs). Type I IFNs play an important role in the development, progression and clinical manifestations of SLE.1 Beyond ICs, neutrophil extracellular traps (NETs), a meshwork of chromatin fibres decorated with antimicrobial proteins, are a more recently described promoter of type I IFN production.2–4
Some patients with SLE have an impaired ability to degrade NETs,5 ,6 a fluctuating phenotype that correlates with glomerulonephritis and hypocomplementemia.7 Further, SLE neutrophils display increased propensity to spontaneously release NETs.2–4 As mechanisms of NET release begin to emerge,8 specific components of lupus NETs, such as cathelicidin/LL-37, have been shown to stimulate both pDCs and macrophages.3 ,9 Vascular and organ damage may also be attributable to NETs in human SLE4 and murine models.10 ,11
Deiminated histones are an important NET component, and peptidylarginine deiminase (PAD)-4 plays a fundamental role in NET formation. Indeed, PAD4−/− mice do not form NETs,12 ,13 and chemical inhibition of PAD4 abrogates NET formation.14 We recently showed that treatment of lupus-prone mice with a PAD inhibitor modulates autoimmune responses and significantly protects against NET-mediated vascular damage.11 These studies were in New Zealand Mixed 2328 (NZM) mice, a strain that manifests female-predominant proliferative glomerulonephritis and strong type I IFN dependence.15 However, not all features of human lupus are replicated in the New Zealand models as these mice do not develop autoantibodies to RNA-containing complexes or spontaneous skin lesions.16
MRL/lpr mice have a spontaneous FAS mutation that dramatically accelerates the lupus phenotype, with massive lymphadenopathy, skin lesions and proliferative glomerulonephritis in sex-independent fashion.16 Some studies have suggested that the MRL/lpr model is not dependent on type I IFNs for development of SLE,17 although that assertion has been called into question.18 A recent study attempted to inhibit NET formation in the MRL/lpr model10 by taking advantage of the fact that NADPH oxidase and reactive oxygen species (ROS) generation are necessary for NET formation in some contexts.19 ,20 Introducing a defective NOX2 (NADPH oxidase) gene into MRL/lpr led to acceleration of the lupus phenotype, especially in terms of nephritis.10 Heterozygous female mice with complete NOX2 deficiency in 50% of neutrophils also had exacerbated lupus, consistent with a protective effect for NOX2 in MRL/lpr.10
Here, we test PAD inhibition in the MRL/lpr model. Our reasoning was that because PAD4 functions downstream of ROS generation during NET formation,14 some important functions of neutrophils may be preserved with PAD inhibition compared with NOX2 mutation. Indeed, NOX2 mutation in its own right has been anecdotally associated with SLE in humans.21 ,22 It should therefore not be assumed that all strategies for preventing NET formation will yield equivalent results. Indeed, in this study, we found an overall protective profile in MRL/lpr mice with two different PAD inhibitors, arguing for a continued exploration of anti-NET therapy in SLE.
Methods
Synthesis of PAD inhibitors
Cl-amidine was synthesised as described.23 BB-Cl-amidine was synthesised as described in online supplementary methods.
BB-Cl-amidine inhibitor characterisation
Inhibitor potency and selectivity were evaluated for PADs 1–4.24 ,25 Cell growth inhibition was evaluated by the XTT assay. Stability was evaluated using a murine hepatic microsome stability assay.26 Pharmacokinetic studies were as previously described.27
Mice and drug treatment
PAD inhibitors were dissolved with 25% DMSO in PBS. MRL/lpr mice were treated with Cl-amidine (10 mg/kg/day), BB-Cl-amidine (1 mg/kg/day) or vehicle by daily subcutaneous injection, beginning at 8 weeks of age until euthanasia at 14 weeks. All mice survived to the end of treatment.
ELISAs and quantitative PCR
Commercial ELISAs for murine anti-ds DNA and total IgG (Alpha Diagnostic), in-house ELISA for anti-cathelicidin-related antimicrobial peptide (CRAMP), RNA isolation and quantitative PCR were performed as described.11
Assessment of endothelium-dependent vasorelaxation and histopathology
Endothelial function studies were performed as reported by us.11 Immunofluorescence staining for kidney IgG and C3 was performed on frozen sections as described.11 Skin and kidney frozen sections were stained with rabbit polyclonal anti-myeloperoxidase (MPO) (Dako) and rat monoclonal anti-Ly-6G (eBioscience). For kidney histopathology, formalin-fixed sections were scored in a blinded manner as described.11 Formalin-fixed kidney and skin sections were also stained with anti-Mx1 (Proteintech).
Statistical analysis
Unless otherwise indicated, statistical analysis was performed using Student's t test in GraphPad Prism software V.6.
Results
MRL/lpr mice display enhanced NET formation and vascular damage
Treatment of MRL/MpJ (control) neutrophils with MRL/lpr serum stimulated NET release compared with treatment with control serum (figure 1A, left). To study this in a serum-independent context, we tested for spontaneous NET release from control and MRL/lpr neutrophils isolated from mice at 14 weeks of age. At this age, MRL/lpr mice have detectable anti-dsDNA autoantibodies, and variably have proteinuria, but are not yet clinically ill. Indeed, under serum-free conditions, MRL/lpr neutrophils spontaneously released more NETs compared with control neutrophils (figure 1A, right). NETs produced by MRL/lpr neutrophils contained proteins previously demonstrated in NETs such as citrullinated histone H3 (figure 1B), MPO and neutrophil elastase (data not shown).14 In contrast to MRL/lpr, B6.lpr mice, which have the same FAS mutation, but a significantly weaker lupus phenotype,28 ,29 demonstrated no difference in NET release under the same conditions (see online supplementary figure S1).

MRL/lpr mice have enhanced neutrophil extracellular trap (NET) formation and vascular damage compared with controls. (A) In the left panel, bone marrow neutrophils were prepared from control MRL/MpJ mice (MRL) and cultured with 10% serum from the indicated 14-week-old mice for 4 h. To the right, bone marrow neutrophils were prepared from the indicated 14-week-old mice and cultured under serum-free conditions for 4 h. NET formation was quantified by fluorescence microscopy. (B) Representative image demonstrating that MRL/lpr NETs contain citrullinated histone H3. DNA is stained blue and citrullinated histone H3 green. The scale bar represents 10 μ. (C) Autoantibodies to cathelicidin-related antimicrobial peptide (CRAMP) were quantified in the serum of the indicated 14-week-old mice. OD index normalises absorbance to the mean value for MRL controls. (D) Aortic rings were isolated from the indicated 14-week-old mice, and acetylcholine-dependent relaxation was determined as described in 'Methods'. N=5 mice for all experiments. Mean±SEM are plotted; *p<0.05; **p<0.01; ***p<0.001.
CRAMP, the murine orthologue of human LL-37, is found in NETs and has important immunostimulatory properties.3 ,30 We previously showed that NZM mice form autoantibodies to CRAMP, and to NETs in general.11 Similarly, compared with MRL control mice, 14-week-old MRL/lpr mice displayed elevated titres of anti-CRAMP autoantibodies (figure 1C).
Given evidence that NETs contribute to vascular damage in SLE,4 ,31 we asked whether the endothelial dysfunction inherent to New Zealand models might also be present in MRL/lpr mice.32 Indeed, 14-week-old MRL/lpr mice showed significantly impaired endothelium-dependent vasorelaxation compared with controls (figure 1D). Overall, these experiments demonstrate accelerated NET release, autoantibody formation to NET components and impaired vascular function in MRL/lpr mice.
Synthesis and characterisation of BB-Cl-amidine, a novel PAD inhibitor
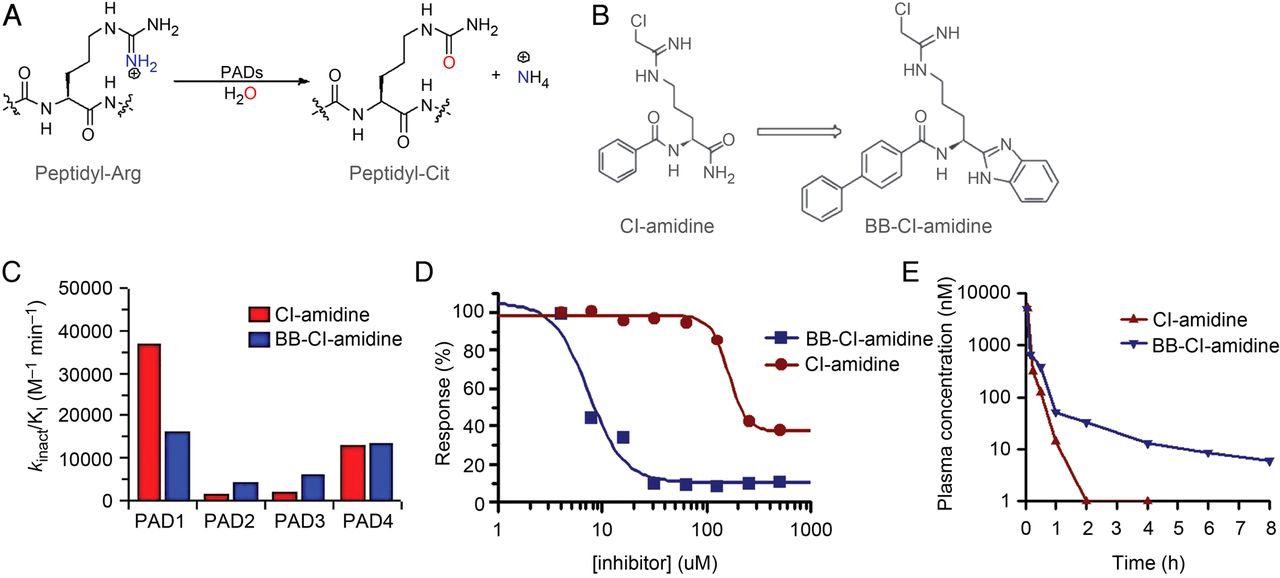
As part of a programme to develop PAD inhibitors with improved cellular bioavailability (figure 2A), we synthesised BB-Cl-amidine. This compound retains the critical elements of Cl-amidine (ie, the reactive chloroacetamidine warhead and sidechain length), but possesses a C-terminal benzimidazole designed to limit proteolysis of the C-terminal amide. Further, the N-terminal benzoyl group is replaced with a biphenyl moiety, which was incorporated to increase hydrophobicity and thereby facilitate cellular uptake (figure 2B). Cl-amidine and BB-Cl-amidine show similar in vitro potencies and selectivities (figure 2C); however, the cellular potency of BB-Cl-amidine is increased by more than 20-fold, evidenced by decreased EC50 values obtained from viability studies with U2OS osteosarcoma cells (8.8±0.6 µM vs >200 µM for Cl-amidine) (figure 2D).33 The enhanced cellular activity is likely due to increased uptake because Cl-amidine and BB-Cl-amidine show similar stabilities in liver microsome stability assays (see online supplementary figure S2), which possess many of the enzymes responsible for drug metabolism in vivo and are a common initial predictor of drug clearance properties. Additionally, we examined the pharmacokinetics of BB-Cl-amidine and found that it has a significantly longer in vivo half-life than Cl-amidine (1.75 h vs ∼15 min) (figure 2E).

Synthesis and characterisation of BB-Cl-amidine. (A) Peptidylarginine deiminase (PAD) reaction mechanism. PADs catalyse the hydrolysis of peptidyl-arginine to form peptidyl-citrulline. (B) BB-Cl-amidine is a C-terminal bioisostere of Cl-amidine. The Cl-amidine structure is based on the structure of benzoyl arginine amide, a small molecule PAD substrate. In BB-Cl-amidine, the C-terminus is replaced by a benzimidazole to prevent proteolysis of the C-terminal amide, and the N-terminal benzoyl group is replaced by a biphenyl moiety to increase hydrophobicity and enhance cellular uptake. (C) Potency and selectivity profiles. The potency (kinact/KI) of Cl-amidine and BB-Cl-amidine for PADs 1 to 4 is depicted graphically. kinact/KI is used because it is the best measure of the potency of an irreversible inhibitor. (D) EC50 of Cl-amidine and BB-Cl-amidine in U2OS cells was assessed. Human osteosarcoma (U2OS) cells were treated with various concentrations of inhibitor for 72 h. Cell viability was measured using the XTT assay. (E) Cl-amidine (10 mg/kg) and BB-Cl-amidine (1 mg/kg) were injected into C57BL/6 mice. Plasma levels were determined by LC-MS/MS.
PAD inhibitors block NET formation, but not H2O2 production
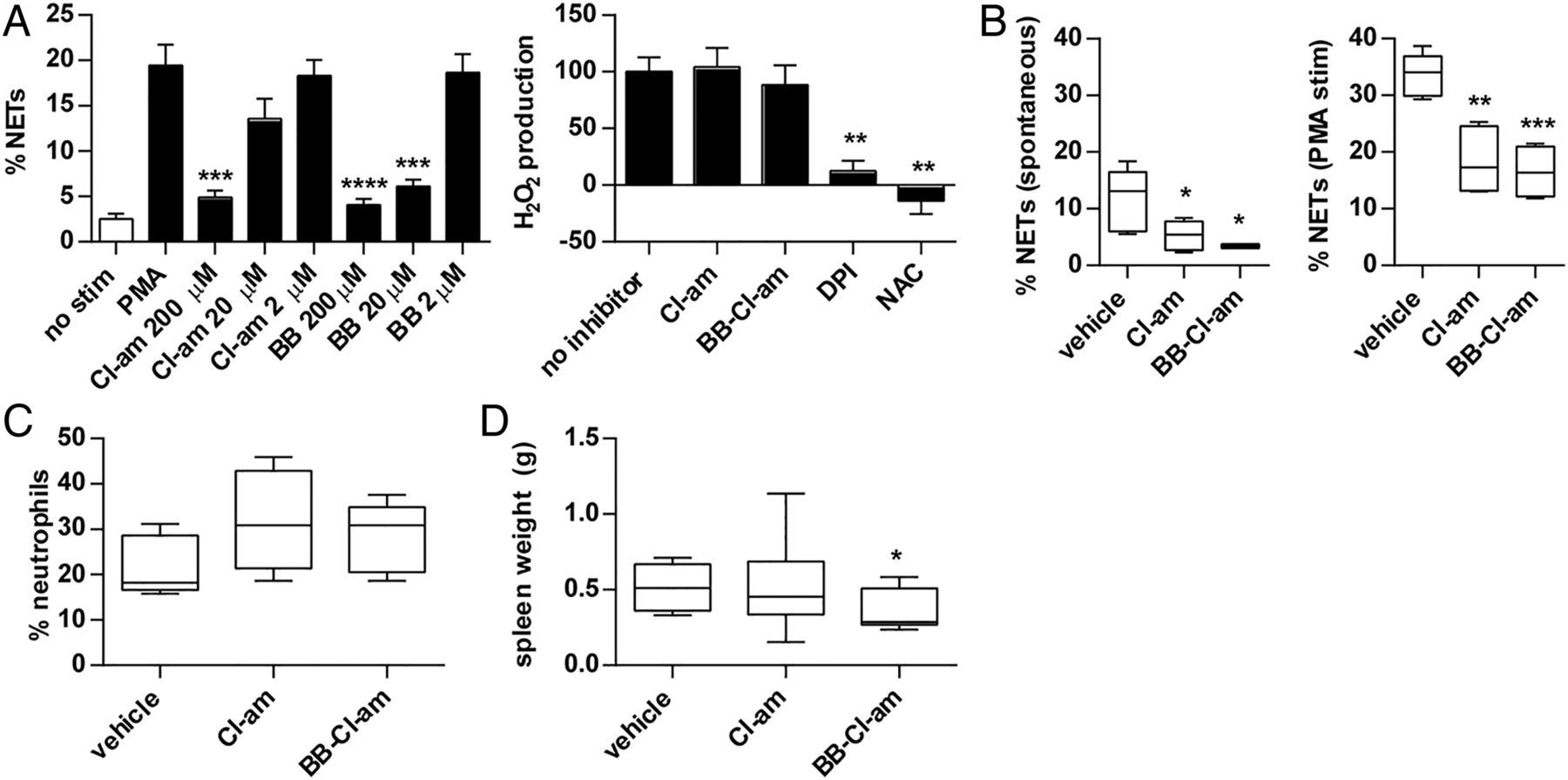
NETs contain deiminated histones and histone deimination by PAD4 plays a fundamental role in NET formation.12–14 We recently showed that the PAD inhibitor Cl-amidine prevents NET formation by NZM lupus neutrophils, in vitro and in vivo.11 Here, we found that Cl-amidine and BB-Cl-amidine significantly inhibited NET formation by MRL/lpr neutrophils (figure 3A, left), while neutrophil H2O2 production was not inhibited by either compound (figure 3A, right). While Cl-amidine was reported to enhance apoptosis in a lymphoma cell line in vitro,34 we did not observe apoptosis induction in freshly isolated splenocytes treated with doses of Cl-amidine that effectively prevent NET formation (see online supplementary figure S3). In contrast, BB-Cl-amidine did induce apoptosis in this system (see online supplementary figure S3). Overall, these data suggest that PAD inhibition impairs the ability of neutrophils to form NETs, without blocking ROS generation through the NADPH oxidase pathway.

Peptidylarginine deiminase inhibition prevents neutrophil extracellular trap (NET) formation when studied with in vitro and ex vivo models. A, Bone marrow neutrophils were prepared from 8-week-old MRL/lpr mice and stimulated with Phorbol 12-myristate 13-acetate (PMA) for 4 h (all black bars received PMA stimulation). NET formation was quantified by fluorescence microscopy. For some samples, neutrophils were pretreated with either Cl-amidine (Cl-am) or BB-Cl-amidine (BB) at the indicated concentrations. To the right, MRL/lpr neutrophils were stimulated for 1 h with PMA in the presence of inhibitors as indicated. The no-inhibitor condition was arbitrarily set at 100% H2O2 production, after subtracting for background H2O2 production from unstimulated cells. DPI, diphenyleneiodonium. NAC, n-acetyl cysteine. The data represent three independent experiments; mean±SEM are plotted. (B) MRL/lpr mice were treated with either Cl-amidine (Cl-am; 10 mg/kg/day) or BB-Cl-amidine (BB-Cl-am; 1 mg/kg/day) by daily subcutaneous injection from 8 to 14 weeks of age. Bone marrow neutrophils were prepared from 14-week-old mice and cultured for 4–6 h, assaying both serum-free spontaneous NET formation (left panel) and PMA-stimulated NET formation (right panel). (C) Circulating neutrophils were determined in the same 14-week-old mice and are plotted as a percentage of total nucleated cells. (D) Spleen weight was measured in the same 14-week-old mice. Boxes represent the median, 25th centile and 75th centile, while whiskers delineate the minimum and maximum values. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
Treatment of MRL/lpr mice with PAD inhibitors prevents ex vivo NET formation
MRL/lpr mice were treated daily with either Cl-amidine or BB-Cl-amidine for 6 weeks. Cl-amidine dosing was based on our previous experience in NZM mice,11 while the BB-Cl-amidine dose was 10-fold lower given the above in vitro data (figure 3A). In the presence of Cl-amidine or BB-Cl-amidine, both spontaneous NET formation and Phorbol 12-myristate 13-acetate (PMA)-stimulated NET formation were reduced (figure 3B). There was a trend towards more circulating neutrophils with PAD inhibition, although this was not statistically significant (figure 3C). Lymphocyte and platelet counts, haematocrit and splenic DCs and T cells were not significantly different between the groups (see online supplementary figure S4 and data not shown). Overall, this demonstrates that ex vivo NET formation is reduced when mice are treated with PAD inhibitors, without significantly altering the number of circulating neutrophils or other immune parameters.
MRL/lpr mice develop marked splenomegaly,35 and there was a subtle reduction in spleen size with BB-Cl-amidine, but not Cl-amidine treatment (figure 3D). Neither group showed a significant difference in spleen architecture, nor was there a significant difference in body weight (data not shown). Further, anti-dsDNA levels were not significantly different between groups at either 11 or 14 weeks of age, although, reminiscent of what we observed in the NZM model,11 there was a trend towards increased circulating anti-dsDNA with PAD inhibition (see online supplementary figure S5). Similarly, for anti-CRAMP, PAD inhibition resulted in significantly increased levels at the 14-week time point (see online supplementary figure S6). In contrast, total IgG levels did not differ between groups (see online supplementary figure S7). In summary, treatment with BB-Cl-amidine subtly reduces splenomegaly in MRL/lpr mice, while there is a trend towards increased circulating levels of anti-NET antibodies with PAD inhibitor treatment. However, neither PAD inhibitor affected body weight or total IgG levels.
PAD inhibition improves vascular function and reduces the IFN signature in MRL/lpr mice
Having shown that MRL/lpr mice have evidence of endothelial dysfunction (figure 1D), we tested whether this dysfunction would be abrogated with systemic administration of PAD inhibitors. Indeed, treatment with both Cl-amidine and BB-Cl-amidine significantly improved endothelium-dependent vasorelaxation (figure 4A). Importantly, neither Cl-amidine nor BB-Cl-amidine altered the contraction profile when added directly to aortic rings in vitro (data not shown), arguing against either compound having a direct effect on endothelial cells.
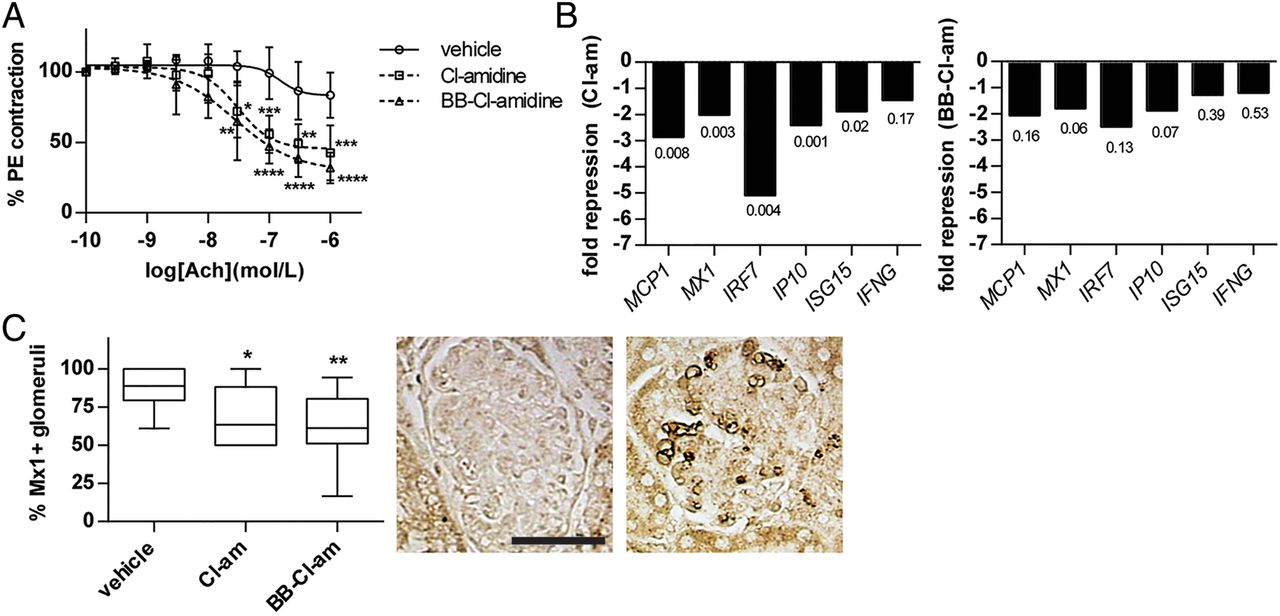
Peptidylarginine deiminase (PAD) inhibition improves vascular function, and decreases interferon (IFN) signature in bone marrow and kidney. MRL/lpr mice were treated with either Cl-amidine (Cl-am; 10 mg/kg/day) or BB-Cl-amidine (BB-Cl-am; 1 mg/kg/day) by daily subcutaneous injection from 8 to 14 weeks of age. (A) At 14 weeks of age, aortic rings were isolated from the indicated groups and acetylcholine-dependent relaxation was determined as described in 'Methods'. (B) At 14 weeks of age, RNA was prepared from total bone marrow and gene expression was determined by quantitative PCR for several type I IFN-regulated genes. The data are expressed as fold change for PAD inhibitor-treated mice, relative to vehicle-treated mice. P values are indicated. (C) At 14 weeks of age, kidneys were harvested, fixed in formalin and stained for Mx1 protein expression (brown). Glomeruli were scored as positive or negative for Mx1, with a minimum of 10 glomeruli counted per mouse. Boxes represent the median, 25th centile and 75th centile, while whiskers delineate the minimum and maximum values. Representative negative (left) and positive (right) staining is shown. Positive staining especially highlights capillary loops. For all experiments, n=10 per group; *p<0.05; **p<0.01; ***p<0.001.
Enhanced type I IFN activity has been associated with the development of endothelial dysfunction in SLE.1 ,31 ,36 Given evidence that NETs are potent stimulators of type I IFNs by pDCs,2–4 a phenotype that we confirmed in vitro for MRL/lpr pDCs (see online supplementary figure S8), we asked whether inhibition of NET formation with PAD inhibitors might downregulate type I IFN-responsive genes (IRGs). RNA isolated from total bone marrow, a compartment containing endothelial progenitors as well as neutrophils prone to spontaneous NET release,11 demonstrated significant downregulation of several IRGs in the Cl-amidine treatment group (figure 4B), as well as when data from the two PAD inhibitors were pooled (data not shown). The BB-Cl-amidine group also showed a strong trend towards downregulation of IRGs (figure 4B). In support of these findings, we detected less MX1 protein in the kidneys of PAD inhibitor-treated mice (figure 4C). Overall, these findings demonstrate improved endothelium-dependent vasorelaxation and decreased expression of IRGs in response to PAD inhibition.
PAD inhibition decreases IC deposition in MRL/lpr kidneys
There was decreased IC deposition, as assessed by IgG and C3 deposition, in MRL/lpr kidneys following PAD inhibition (figure 5A and B). In terms of the inflammatory response, when data for the two inhibitors were pooled, there was a significant reduction in interstitial inflammation (figure 5C); there was also a reduction in overlapping DNA and MPO staining (consistent with NETs) in the glomeruli of treated mice (see online supplementary figure S9). There was a trend towards reduced overall glomerular activity with PAD inhibition, which did not reach statistical significance (figure 5C). When the data for both PAD inhibitors were pooled, there was a significant decrease in urine albumin/creatinine ratio compared with vehicle-treated mice (figure 5D). Overall, these results indicate that PAD inhibition improves lupus kidney disease in MRL/lpr mice.

Peptidylarginine deiminase (PAD) inhibition decreases IC deposition in kidneys and reduces proteinuria. MRL/lpr mice were treated with either Cl-amidine (Cl-am; 10 mg/kg/day) or BB-Cl-amidine (BB-Cl-am; 1 mg/kg/day) by daily subcutaneous injection from 8 to 14 weeks of age. (A) Representative anti-IgG staining is shown with a vehicle-treated glomerulus on the left and PAD inhibitor-treated on the right. Quantification used a scoring system ranging from 0 to 3+ as described in 'Methods'. (B) Representative anti-C3 staining is shown with a vehicle-treated glomerulus on the left and PAD inhibitor-treated on the right. Quantification used a scoring system ranging from 0 to 3+ as described in 'Methods'. (C) Periodic acid-Schiff (PAS)-stained sections were scored for activity (activ.) and chronicity (chron.) as described in 'Methods'; there were no statistical differences in the left graph. Interstitial inflammation was also quantified, and pooled data for Cl-am and BB-Cl-am is denoted as ‘PAD inh.’ (D) Proteinuria was determined at 14 weeks of age by calculating the ratio of albumin to creatinine in the urine. Pooled data for Cl-am and BB-Cl-am are denoted as ‘PAD inh.’ For all experiments, n=10 per group; *p<0.05; **p<0.01; ***p<0.001. Boxes represent the median, 25th centile and 75th centile, while whiskers delineate the minimum and maximum values.
PAD inhibition improves skin involvement in MRL/lpr mice
Untreated MRL/lpr mice lost whiskers and developed patches of fur loss on the muzzle by 14 weeks of age (figure 6A). This phenotype has been reported as a manifestation of cutaneous lupus.18 ,37 Dorsal neck lesions, which have been described in MRL/lpr mice at 20 weeks,38 were not present at the time points tested here. Treatment with either Cl-amidine or BB-Cl-amidine significantly improved muzzle alopecia, in many cases preventing it entirely (figure 6A). Overlapping DNA and MPO staining, consistent with NETs, was easily detectable in the dermis of these patches (figure 6B) and was significantly reduced by PAD inhibitors (figure 6B). Consistent with the idea that NETs are inducers of type I IFNs, Mx1-positive cells were also significantly reduced in the dermis of PAD inhibitor-treated mice (figure 6C). These observations suggest that PAD inhibitors not only reduce NET formation and IFN production in MRL/lpr skin, but can also clinically mitigate alopecia.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Peptidylarginine deiminase (PAD) inhibition improves skin involvement in MRL/lpr mice. MRL/lpr mice were treated with either Cl-amidine (Cl-am; 10 mg/kg/day) or BB-Cl-amidine (BB-Cl-am; 1 mg/kg/day) by daily subcutaneous injection from 8 to 14 weeks of age. (A) Representative area of alopecia in a 14-week-old mouse treated with vehicle. To the right is quantification of lesion circumference in the presence or absence of PAD inhibition. (B) The area of alopecia (or the corresponding area in mice without visible lesions) was sampled and frozen sections were prepared. Dermal neutrophil extracellular trap were determined as areas of MPO-DNA overlap. At least five 400× fields were counted per mouse. Representative staining is shown to the right with DNA in blue and MPO in green; for orientation, the epidermis is marked with an ‘E.’ Scale bar=25 μ. (C) Formalin-fixed, paraffin-embedded sections were prepared, and Mx1-positive cells within the dermis were quantified. Representative vehicle-treated (left) and PAD inhibitor-treated (right) sections are shown. Mx1 protein is stained brown. The secondary antibody reacted non-specifically with the epidermis (E) as well as hair follicles (HF). Scale bar=50 μ. For all experiments, n=10 per group. Boxes represent the median, 25th centile and 75th centile, while whiskers delineate the minimum and maximum values; *p<0.05; **p<0.01; ***p<0.001.
Discussion
MRL/lpr mice replicate many of the features we previously described in the NZM model,11 including an enhanced rate of spontaneous NET formation and autoantibodies to the NET constituent CRAMP, considered potent stimulators of type I IFN production by pDCs.3 ,30 We also demonstrate that MRL/lpr mice have significant evidence of endothelial dysfunction. We attribute this dysfunction to the lupus phenotype, as our previous work in B6.lpr, a strain with only mild lupus-like disease, did not demonstrate similar dysfunction.32
Both Cl-amidine and the novel PAD inhibitor BB-Cl-amidine effectively inhibited NET formation in vitro, as well as when neutrophils from treated mice were studied ex vivo. Overall, chemical PAD inhibition seems to be relatively specific for NET formation and, importantly, did not prevent the formation of ROS (suggesting that the NADPH oxidase system is intact in the studies presented here). Further, circulating lymphocytes, neutrophils and total IgG levels were not altered by PAD inhibition, nor was there an effect on splenic DCs or T cells. In our previous work, Cl-amidine did not regulate B- and T-lymphocyte activation in vitro,11 and cell depletion experiments have strongly suggested that Cl-amidine's ability to protect against vascular damage is primarily through inhibition of NET release.39 These observations are generally supported by the work of others, with Cl-amidine having no detectable impact on immune cell numbers in inflammatory arthritis models.40
Similar to our previous work in the NZM model,11 there was a trend towards increased anti-dsDNA and anti-CRAMP with PAD inhibition, with no difference observed for total IgG, arguing that this trend is relatively specific for anti-NET antibodies. Anti-dsDNA titres inversely correlated with IgG deposition in kidneys, leading us to hypothesise that with less NET release, there is decreased IC formation, and consequently less opportunity for anti-NET antibodies to deposit in tissues. This is in line with both our previous work11 and the work of others.41 ,42 An alternative explanation is that increased in vivo apoptosis (as suggested for BB-Cl-amidine in online supplementary figure S3) leads to the release of more autoantigens and ultimately an increased autoantibody response. This hypothesis is supported by previous work in MRL/lpr mice.43
Treatment with Cl-amidine and BB-Cl-amidine markedly improved endothelium-dependent vasorelaxation. This is relevant as endothelial dysfunction is highly prevalent in lupus patients, predates atherosclerotic clinical disease and correlates with elevated type I IFN signature.1 ,31 Here, we found a robust suppression of multiple IRGs in the bone marrow of treated MRL/lpr mice, a compartment where endothelial progenitors arise.11 This experimentally supports the in vitro work of others in human cells, where NETs have been shown to be potent activators of type I IFN synthesis by pDCs.2–4 In support of the RNA data, less Mx1 protein was detected in kidneys and skin of treated mice. This is noteworthy as the MX1 gene is considered highly type I IFN specific.44 In the kidneys, a significant positive correlation between IgG deposition and Mx1 staining was found, supporting our hypothesis that NETs and anti-NET antibodies are important stimulators of type I IFN production. While we also tested splenocytes for an enhanced IFN signature, there was no difference with PAD inhibition (data not shown), probably explained by the lack of netting neutrophils and tilting toward double-negative T cells in this compartment.16 ,45
PAD inhibition decreased renal inflammation, IC deposition and proteinuria. For this study, given the substantial amount of inhibitor required for daily injection, we tested a relatively early time point, at which not all vehicle-treated mice had yet developed proteinuria. Also, this 14-week time point was such that all mice survived. Follow-up studies should characterise proteinuria and survival at later time points and address potential benefits of PAD inhibition once nephritis already developed.
Of particular interest is the improvement in skin disease with PAD inhibition, a clinical manifestation not previously targeted with these compounds. In human SLE, systemic and skin IFN signatures correlate with severity of cutaneous manifestations.46 ,47 A model of induced cutaneous lupus in NZB/W F1 mice results in NETs and pDC recruitment to the dermis, leading to enhanced IRGs and prolonged rash.48 Here, we showed a marked improvement in MRL/lpr muzzle lesions with both PAD inhibitors, correlating with diminished MPO/DNA complexes and Mx1-positive cells in the dermis. This raises the possibility of exploring topical or systemic PAD inhibitors for the treatment of cutaneous lupus.
The apparent differences between NOX2 mutation and PAD inhibition in the MRL/lpr model are intriguing.10 An important point is that PAD4 probably functions downstream of ROS generation during NET formation14; some neutrophil functionality may therefore be preserved with PAD inhibition compared with NOX2 mutation. Further, NOX2 mutation has been associated with a proinflammatory state in humans,49 ,50 and has even been posited to directly predispose to SLE.21 ,22 NOX2 mutation also appears to alter both inflammasome activation51 and antigen presentation52 in ways that could exacerbate SLE.53 Further studies will be necessary, and ideally other forms of NET inhibition tested, in the MRL/lpr model to resolve these discrepancies.
In summary, NET abrogation through PAD inhibition effectively mitigates vascular, kidney and skin disease in the MRL/lpr model of lupus, indicating that this therapeutic strategy can be effective in different models of lupus. These observations further support the concept that inhibition of aberrant NET formation should be further explored in SLE as a therapeutic strategy.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
JSK and VS contributed equally.
Contributors JSK designed and performed experiments and drafted the manuscript. VS performed experiments and drafted the manuscript. AAO, SY and CKS performed experiments. WZ performed experiments and contributed to statistical analysis.. JBH analysed data. PRT designed experiments and drafted the manuscript. MJK conceived and designed study and drafted the manuscript.
Funding Microscopy was performed in the Center for Live Cell Imaging (CLCI) at the University of Michigan and at the IRP at NIAMS. JSK was supported by a Rheumatology Research Foundation Rheumatology Scientist Development Award. This work was supported by the National Institutes of Health (NIH) through PHS grant HL088419 (to MJK) and by the Intramural Research Program at NIAMS. PRT is supported by NIH grants GM079357 and CA151304.
Competing interests None.
Provenance and peer review Not commissioned; externally peer reviewed.