Article Text

Abstract
Objectives High levels of vascular endothelial growth factor (VEGF), a key angiogenic factor, are present in patients with systemic sclerosis (SSc), but its role in the pathogenesis of fibrosis and its contribution to the disturbed angiogenesis of SSc remains hypothetical.
Methods Mono (+/−) and double (+/+) VEGF transgenic (tg) mice and their wildtype (wt) controls were analysed. The bleomycin model was applied to VEGF tg mice to evaluate effects of VEGF under proinflammatory conditions. Additionally, tight skin (TSK) 1/VEGF+/+ mice were generated to mimic later non-inflammatory stages of SSc.
Results VEGF+/+, but not VEGF+/− tg mice, spontaneously developed significant skin fibrosis, indicating profibrotic effect of VEGF in a gene-dosing manner. In the proinflammatory bleomycin model, the profibrotic effect became more pronounced with induction of skin fibrosis in VEGF+/− tg mice and even more enhanced fibrosis in VEGF+/+ tg mice. Analysis in TSK1/VEGF+/+ mice showed similar profibrotic effects of VEGF also under non-inflammatory in vivo conditions. In vitro analysis revealed that VEGF is able to directly induce collagen synthesis in dermal fibroblasts. Additionally, there was an inverse gene-dosing effect on the efficacy of angiogenesis in that a higher number of microvessels was observed in VEGF+/− tg mice than in VEGF+/+ tg mice.
Conclusions These data provide the first evidence for VEGF as a novel molecular link between fibrosis and vasculopathy in the pathogenesis of SSc. They suggest that high levels of VEGF potently induce fibrosis in inflammatory and non-inflammatory stages, and also contribute to the relatively insufficient angiogenesis characteristic for SSc.
- Systemic Sclerosis
- Fibroblasts
- Inflammation
Statistics from Altmetric.com
Vascular damage is considered one of the earliest pathogenic events in systemic sclerosis (SSc), since vascular manifestations such as Raynaud's phenomenon and morphological changes on nailfold capillaroscopy often precede the onset of other clinical manifestations.1 The identification of pathways linking vasculopathy to organ fibrosis might thus provide important insights into early disease mechanisms and allow early targeted intervention for both fibrotic and vascular events. However, the underlying mechanisms linking these two major pathways remain largely hypothetical.2
Vascular endothelial growth factor (VEGF) is a potent proangiogenic molecule. However, the proangiogenic effects depend on a tight control of its temporal and spatial expression patterns.3 Accordingly, several studies suggested that chronic, uncontrolled overexpression of VEGF results in a disturbed morphology of blood vessels reminding of the microvascular pathology seen in SSc.4–6
In SSc, levels of VEGF are strongly increased in sera and skin biopsies as compared with healthy controls.7 ,8 They are detectable even in patients with very early disease and remain chronically elevated across different disease stages. The upregulation of VEGF is especially pronounced in diffuse cutaneous SSc.7 SSc patients in very early prefibrotic disease stages, who later developed skin fibrosis, had higher levels of VEGF compared with those in whom no cutaneous involvement occurred at follow-up. These data indicate a possible role of VEGF in the pathogenesis of fibrosis.7
The aim of our study was therefore to analyse the effects of VEGF on fibrosis and angiogenesis in different animal models of SSc using VEGF transgenic mice9 in which the constitutive expression of the VEGF transgene reflects the high tissue levels of VEGF in SSc skin.
Methods
Additional information on methods is provided in the online supplementary data.
Patients and biopsy specimens
Skin fibroblasts were derived from skin biopsies from SSc patients fulfilling the LeRoy criteria10 (see online supplementary table S1) and from healthy donors. Skin fibroblasts were expanded by outgrowth culture.11 Written informed consent was obtained from all patients, and the study was approved by the local ethics committee.
Cell cultures, reagents and stimulation assays
After 24 h of serum reduction, SSc and normal skin fibroblasts were stimulated for 24 and 48 h with recombinant VEGF-A165 (R&D Systems, Abingdon, UK) at 10, 20 and 100 ng/mL. Controls were exposed to equivalent dosages of the carrier protein BSA.12 For the analysis of VEGF receptor (VEGFR), normal and SSc skin fibroblasts were compared with human umbilical vein endothelial cells (HUVECs).
Real-time RT-PCR
Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Hombrechtikon, Switzerland) and reverse transcribed into complementary DNA with random hexamers.11 Gene expression was quantified by SYBR Green real-time PCR using the ABI Prism 7700 Sequence Detection System (PE Applied Biosystems, Rotkreuz, Switzerland). Specific primer pairs for human procollagen I and III were designed with Primer 3 software and synthesised from Microsynth (Balgach, Switzerland). For the detection of VEGFR RNA, specific gene expression assays were used (Applied Biosystems, Basel, Switzerland).
Measurement of collagen proteins
To assess the effects of VEGF stimulation in SSc compared with normal fibroblasts, total soluble collagen in cell culture supernatants was quantified using the Sircol collagen assay (Biocolor, Belfast, UK).11
Animal models of SSc
Bleomycin-induced dermal fibrosis in VEGF tg mice as an early inflammatory model of SSc
To investigate the effects of VEGF in an inflammatory model of SSc mono (+/−) (n=9), double (+/+) (n=8) VEGF tg mice9 and wild type (wt) littermates (n=6/9) were treated with bleomycin.11 ,13
In VEGF tg mice, the murine VEGF164 gene, which is the murine equivalent to VEGF-A165, was cloned into a human keratin 14 promoter expression cassette, which had previously been shown to selectively target transgene expression to basal keratinocytes of the skin leading to an increased expression of VEGF in the dermis.9 VEGF tg mice were on the Friend leukemia virus B (FVB) background.
Skin fibrosis was induced in 6-week-old FVB male mice by local intracutaneous injections of 100 µL of bleomycin dissolved in 0.9% NaCl, at a concentration of 0.5 mg/mL, every other day in defined areas of 1.5 cm2 on the upper back for 28 days.11 ,13 Saline-treated VEGF double (+/+) and mono tg (+/−) mice were used as controls for the bleomycin experiment as well as to assess angiogenesis and fibrosis in VEGF tg mice.
Overexpression of VEGF in TSK1 mice as a late-stage, fibrotic model of SSc
To assess the effects of VEGF in a non-inflammatory model, tight skin (TSK) 1/VEGF+/+ mice were generated by crossing VEGF(+/+) tg mice with TSK1 mice.14 Due to a dominant mutation of the fibrillin 1 gene, TSK1 mice develop an increased dermal and especially hypodermal thickness.14 TSK1/VEGF+/+ mice (n=4) were compared with TSK1 (n=5) and wt (pa/pa) mice (n=4).13
In the present study, all mice were analysed at 10 weeks of age.
Histological and immunohistological analysis
Murine skin sections were fixed in 4% formalin, embedded into paraffin and stained with haematoxylin and eosin (HE) and Masson's trichrome staining according to standard protocols. Polyclonal rabbit anti-vWF as a marker of endothelial cells (abcam, Cambridge, UK) and monoclonal mouse anti-α-smooth muscle actin (SMA) as a marker for vascular smooth muscle cells and myofibroblasts (Sigma-Aldrich, Buchs, Switzerland) were used as primary antibodies.
All slides were analysed by two blinded independent examiners. Dermal thickness was analysed at ×100 magnification as previously described.11 In TSK1 mice, hypodermal thickness was determined at ×25 magnification as previously described.15 To determine the number of myofibroblasts as additional parameter of skin fibrosis, pictures at ×100 magnification were taken, and spindle-shaped, α-SMA+ cells were counted.
For the semiquantitative analysis of microvessel density, pictures at ×100 magnification were taken. Then, α-SMA+/vWF+ blood vessels (dermal microvessels) were counted. Blood vessel density was assessed in the dermis except for TSK1 or TSK1/VEGF+/+ mice of which the hypodermis was analysed. Vessel wall thickness of α-SMA+/vWF+ vessels was assessed as a parameter of the proliferative vasculopathy of SSc. Pictures were taken at ×1000 magnification.
For the semiquantitative analysis of inflammatory infiltrates, pictures at ×100 magnification were taken. Then, F4/80+ macrophages (green) and CD3+ lymphocytes (purple) were counted.
Statistical analysis
For statistical analysis, GraphPad Prism software (V.5.01) was employed. Normal distribution of data was examined using the Kolmogorov–Smirnov test. For parametric non-related data, expressed as mean±SEM, the unpaired two-tailed t test was used. Nonparametric non-related data, expressed as median(Q1, Q3), were analysed employing the Mann–Whitney-U test. p Values less than 0.05 were considered statistically significant.
Results
Angiogenesis, vasculopathy and fibrosis in VEGF tg mice
In mice overexpressing VEGF, the number of dermal microvessels (vWF+/α-SMA+) increased spontaneously. This was most pronounced in VEGF+/− tg mice (figure 1B). The proangiogenic effects occurred also in VEGF+/+ tg (figure 1C) as compared with wt mice (figure 1A), but to a significantly lesser extent than in VEGF+/− tg mice. Semiquantitative analysis by blinded examiners confirmed this finding (mean±SEM VEGF+/− tg 21.5±1.3 vs VEGF+/+ tg 15.3±3.1 vs 5.2±1.1/high power field (HPF) in wt mice; p<0.05; figure 1G). Interestingly, we also observed an increase in vessel wall thickness in VEGF tg mice (figure 1D–F+H; VEGF+/− tg 4.2±0.2 µm vs VEGF+/+ tg 3.5±0.3 µm vs 1.9±0.2 µm in wt mice; p<0.05) with α-SMA+ cells in the vessel wall resembling findings of the proliferative vasculopathy in SSc.

Angiogenic effects of VEGF in vivo. The figure shows the immunohistochemical staining of dermal microvessels (vWF+ red, α-smooth muscle actin (SMA)+ blue). In VEGF+/− tg mice (B), microvessel density was most increased compared with wt (A) and VEGF+/+ mice (C). The effect on vessel wall thickness of α-SMA+ (blue) vessels (red arrows) was also most pronounced in VEGF+/− mice (E) compared with wt (D) and VEGF+/+ mice (F). (G+H) display the results of the semiquantitative analysis by blinded examiners. Pictures are representative examples of 8 homozygous, 9 heterozygous VEGF tg mice and their respective wt littermates (n=6/9). Data are expressed as mean±SEM. *indicates p values <0.05.
We next evaluated the effects of VEGF on skin fibrosis. VEGF+/+ tg (figure 2C) mice, but not VEGF+/− tg mice (figure 2B), spontaneously developed skin fibrosis as compared with wt mice (figure 2A) (mean±SEM skin thickness 437.9±7.8 vs 378.6±16.9μm, p<0.05, figure 2G). To exclude the possibility that the increase in skin thickness was due to skin oedema secondary to VEGF-induced vascular permeability, extracellular matrix staining with Masson's trichrome was performed. These analyses confirmed that the increase in skin thickness was due to fibrosis caused by deposition of extracellular matrix proteins (figure 2D–F). Furthermore, the number of α-SMA+ myofibroblasts as an additional marker of fibrosis (figure 1A–C) was significantly increased in VEGF+/+ tg mice versus VEGF+/− tg mice and wt mice, respectively (mean±SEM 7.3±1.1 vs 2.8±0.3 and 2.3±0.2 myofibroblasts/HPF; p<0.05; figure 2H). No differences were found in the number of infiltrating F4/80+ macrophages and CD3+ T lymphocytes in VEGF+/+ and VEGF+/− tg mice versus wt mice indicating effects independent from these cells (see supplementary figure S2).

Profibrotic effects of VEGF in vivo. The figure shows the differences in dermal thickness as analysed by haematoxylin and eosin and Masson's trichrome staining. VEGF+/+ spontaneously developed an increase in dermal thickness (C) compared with wt (A) and VEGF+/− mice (B). The latter did not differ from wt mice. Additional Masson's trichrome staining confirmed that the increase in skin thickness was due to deposition of extracellular matrix proteins and not due to oedema caused by VEGF-induced vascular leakage (D–F). (G+H) displays the results of the semiquantitative analysis of skin thickness and myofibroblast count as measures of fibrosis. Pictures are representative examples of 8 homozygous, 9 heterozygous VEGF tg mice and their respective wt littermates (n=6/9). Data are expressed as mean±SEM. *indicates p values <0.05.
These results show that overexpression of VEGF can by itself spontaneously induce dermal fibrosis in vivo at high doses coincidentally with an induction of angiogenesis and vasculopathy. Interestingly, the effects on angiogenesis were significantly stronger in VEGF mono tg mice than in VEGF double tg mice suggesting that angiogenesis is best achieved with medium levels of VEGF, but not with high levels as in VEGF double tg mice.16
Angiogenesis and aggravated fibrosis/vasculopathy in bleomycin challenged VEGF tg mice
Next, we analysed the effects of VEGF in a proinflammatory environment using the bleomycin skin fibrosis model, which mimics early disease stages of SSc.
First, challenge of VEGF tg mice with bleomycin enhanced the observed effects of VEGF on angiogenesis and vasculopathy. VEGF+/− tg mice (figure 3B/E), as compared with VEGF+/+ tg mice (figure 3C/F) and wt mice (figure 3A/D), showed the most pronounced increase in both the number of microvessels (figure 3G) (mean±SEM 31.8±1.6 vs 19.1±1.4 vs 10.2±0.9/HPF; p<0.05) and vessel wall thickness (figure 3H) (4.0±0.2 µm vs 3.3±0.1 µm vs 2.3±0.2 µm; p≤0.05).
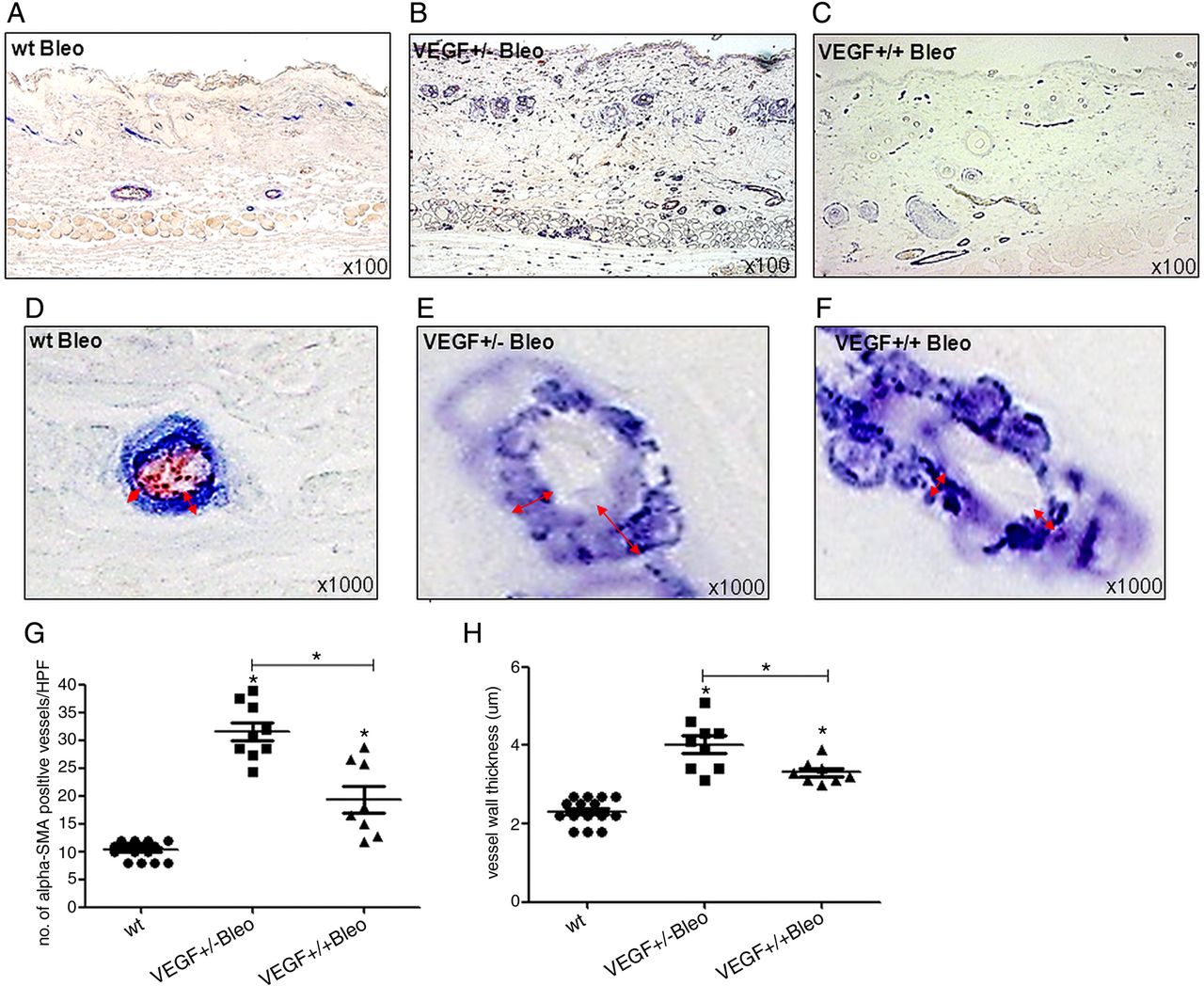
Angiogenic and vasculopathic effects of VEGF in the bleomycin model. The figure shows the immunohistochemical staining of dermal microvessels (vWF+ red, α-smooth muscle actin (SMA)+ blue). In bleomycin challenged VEGF+/− tg mice (B), microvessel density as a marker of angiogenesis was most increased compared with wt (A) and VEGF+/+ mice (C). The effect on vessel wall thickness of α-SMA+ (blue) vessels (red arrows) as a marker of vasculopathy was also most pronounced in VEGF+/− mice (E) compared with wt (D) and VEGF+/+ mice (F). (G+H) display the results of the semiquantitative analysis. Pictures are representative examples of 8 homozygous, 9 heterozygous VEGF tg mice and their respective wt littermates (n=6/9). Data are expressed as mean±SEM. *indicates p values <0.05.
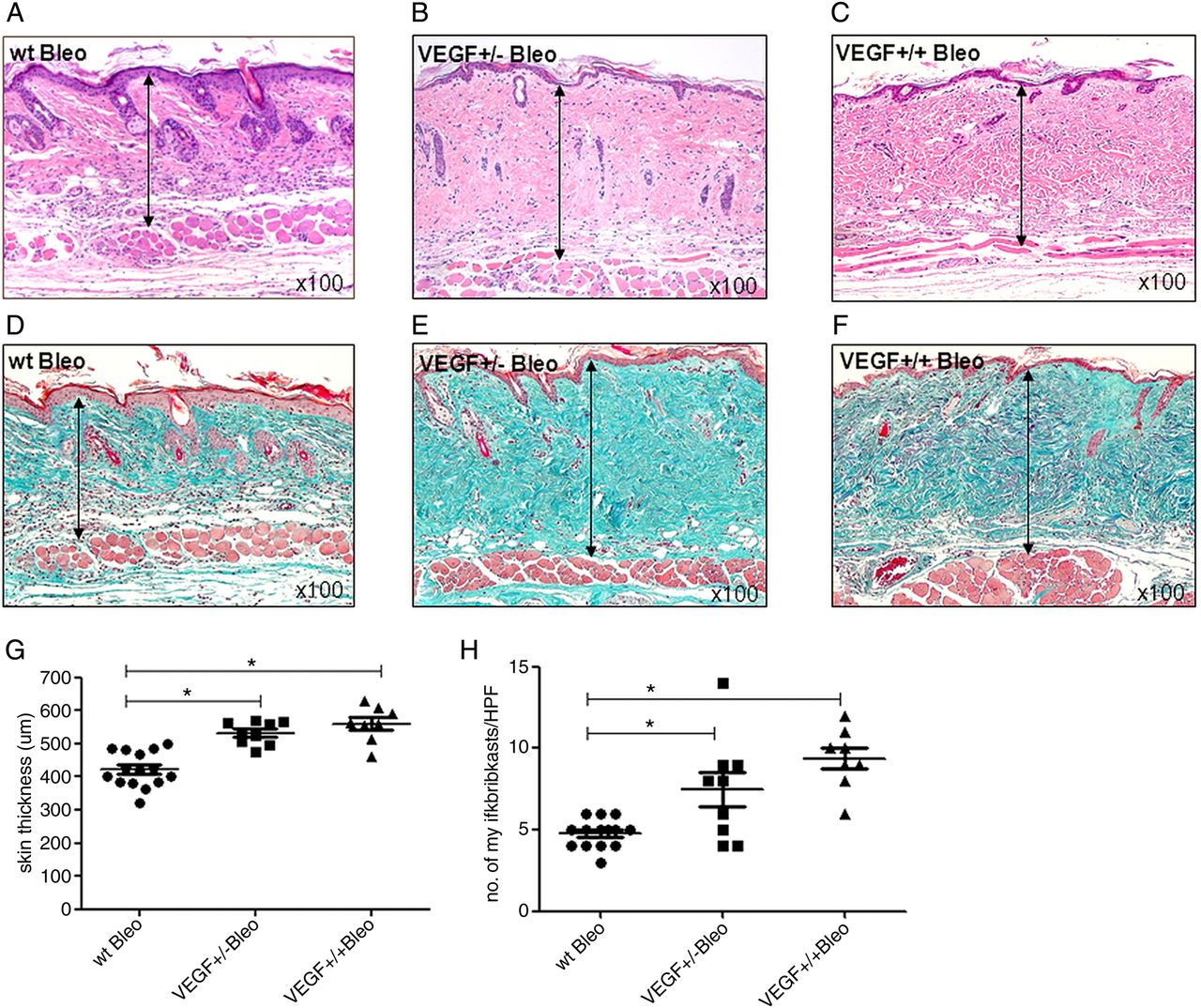
Additionally, compared with wt mice (figure 4A), bleomycin challenge further increased skin thickness in VEGF+/+ tg (figure 4C) and induced skin fibrosis in VEGF+/− tg mice (figure 4B) (mean±SEM skin thickness 559.8±19.0, 531.9±11.5μm; p<0.05; figure 4G), which was also demonstrated by Masson's trichrome stain (figure 4D–F) and by an increase in the number of α-SMA+ myofibroblasts (figure 3A–C) (figure 4H; mean±SEM 9.4±0.7 vs 7.4±1.1 vs 4.7±0.3 myofibroblasts/HPF; p<0.05). Of note, VEGF+/+ tg mice without bleomycin challenge showed the same increase in skin thickness as bleomycin challenged wt mice (figure 4H) indicating a similarly strong profibrotic effect of VEGF itself as with bleomycin.

Profibrotic effects of VEGF are enhanced in the bleomycin model. The figure shows differences in dermal thickness as analysed by haematoxylin and eosin and Masson's trichrome staining. Bleomycin challenged VEGF+/+ mice (C) showed a significant increase in dermal thickness compared with wt mice (A), which was further increased compared with unchallenged VEGF+/+ mice (figure 2C). Other than unchallenged VEGF+/− mice (figure 2B), bleomycin challenged VEGF+/− mice (B) now also developed an increase in dermal thickness. As demonstrated by additional Masson's trichrome staining (D–F), the increase in skin thickness was caused by a deposition of extracellular matrix proteins and not due to oedema secondary to VEGF-induced vascular leakage. (G+H) display the results of the semiquantitative analysis of skin thickness measurements and myofibroblast counts. Pictures are representative examples of 8 homozygous, 9 heterozygous VEGF tg mice and their respective wt littermates (n=6/9). Data are expressed as mean±SEM. *indicates p values <0.05.
In conclusion, these data suggest that VEGF dose-dependently aggravates fibrosis and vasculopathy in early inflammatory stages of SSc. Again, proangiogenic effects were mostly seen with moderate doses of VEGF, but less so with high levels of VEGF.
Worsening of fibrosis and vasculopathy in TSK1/VEGF+/+ mice
We next aimed to analyse whether VEGF is also able to aggravate fibrosis and the proliferative vasculopathy under non-inflammatory conditions. For this approach, we used the TSK1 mouse model of SSc that resembles non-inflammatory, later disease stages and crossed VEGF+/+ tg with TSK1 mice to generate TSK1 mice overexpressing VEGF.
Whereas TSK1 mice (figure 5B) showed a reduced number of dermal microvessels compared with wt mice (figure 5A) (median(Q1,Q3) 2.0(1,2) vs 5.5(3,6)/HPF; p<0.05; figure 5D), in TSK1/VEGF+/+ mice (figure 5J), microvessel density (16.5(11,25) vs 2.0(1,2)/HPF in TSK1 mice; p<0.05; figure 5J) and vessel wall thickness (figure 5D–F) increased (4.9(4,8) vs 2.9(3,4)µm in TSK1 mice; p<0.05; figure 5K).

Angiogenic and profibrotic effects of VEGF in the tight skin (TSK) 1 model. (A–F) show the immunohistochemical staining of dermal microvessels (vWF+ red, α-smooth muscle actin (SMA)+ blue). Compared with wt (A/D) and TSK1 mice (B/E), TSK1/VEGF+/+ mice (C/F) showed an increased number and vessel wall thickness of α-SMA+ (blue) vessels (red arrows) of dermal microvessels. (J+K) show the semiquantitative analysis. (G–I) demonstrate the differences in hypodermal thickness as analysed by haematoxylin and eosin staining. Compared with wt (pa/pa) (G) and TSK1 mice (H), TSK1/VEGF+/+ mice (I) developed an increase in hypodermal thickness that was more pronounced than that of TSK1 mice. (L+M) display the results of the semiquantitative analyses of skin thickness and myofibroblast count as measures of fibrosis. Pictures are representative examples of 4 TSK1/VEGF+/+ mice, 5 TSK1 and 4 wt (pa/pa) mice. Data are expressed as median(Q1,Q3). *indicates p values <0.05.
This was paralleled by an increase in hypodermal thickness in TSK1/VEGF+/+ (figure 5I) compared with TSK1 (figure 5H) and wt mice (figure 5G) (median(Q1,Q3) 690(670,723) vs 470(415,490) vs 105(92,110)μm; p<0.05; figure 5L). These results were confirmed by Masson's trichrome staining (see supplementary figure S1) and by an increase in the number of myofibroblasts (median(Q1,Q3) 46(18,69) 16.0(12,19) vs 4(3,5) myofibroblasts/HPF; p<0.05; figure 5M).
Taken together, these data suggest that the strong profibrotic effects as well as the vasculopathic effects of VEGF were also operative under non-inflammatory conditions. VEGF might thus be an important mediator of fibrosis and vasculopathy not only in early but also in later, non-inflammatory stages of SSc.
VEGF increases collagen production in vitro
Next, we evaluated the profibrotic effects of VEGF in vitro. Stimulation of normal and SSc skin fibroblasts with recombinant VEGF-A dose-dependently enhanced the production of collagens although compared with HUVECs, human skin fibroblasts do not express VEGFRs (see supplementary figure S38 ,12;). In SSc fibroblasts (figure 6B), the raise in procollagen 1A1 and 3A1 mRNA levels was more pronounced than in normal skin fibroblasts (figure 6A) (by median(Q1,Q3) 3.0(2,5) and 6.8(5,11)fold vs 2.5(2,3) and 1.8(1.5,2) fold compared with the respective controls; p<0.05). Induction of collagens peaked after 48 h (data not shown). Similarly, in the Sircol assay (figure 6C), the supernatants of VEGF-stimulated SSc fibroblasts contained more collagen protein than control fibroblasts stimulated with the carrier solution (10 ng/mL VEGF 63.3(54,70), 100 ng/mL VEGF 86.7(65,89) vs 52(46,62) ng/mL). These data suggest direct profibrotic effects of VEGF on dermal fibroblasts as one of the mechanisms contributing to the profibrotic effects of VEGF.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
VEGF induces collagen synthesis in dermal fibroblasts. In SSc (A) and normal skin fibroblasts (B), stimulation with recombinant VEGF-A increased procollagen I and III mRNA. (C) Similarly, in the Sircol assay, the supernatants of VEGF stimulated SSc fibroblasts contained significantly more collagen protein than those of control fibroblasts stimulated with the carrier solution of recombinant VEGF.
Discussion
Our in vivo studies with VEGF tg mice, bleomycin-induced fibrosis and TSK1 mice convincingly showed that VEGF dose-dependently exerts strong profibrotic effects that lead to a chronic fibrotic stage with increased skin thickness at 10 weeks, 3 months and 5 months in VEGF double tg mice16 (for more detailed discussion refer to online supplementary material). These profibrotic effects were accompanied by a vasculopathy with an increase in vessel wall thickness, which is a classical feature of the proliferative microangiopathy seen in SSc. In addition, high levels of VEGF in double tg VEGF mice hampered the proangiogenic effects of VEGF, which is also seen in SSc patients with a chronic and high upregulation of VEGF and insufficient angiogenesis.8 These findings are of potential importance, because they provide a molecular link between fibrosis and vasculopathy, which are two of the major pathogenic pathways leading to clinical manifestations in SSc.
The profibrotic effects of VEGF are likely mediated by multiple mechanisms. It is well established that VEGF attracts VEGFR1 expressing monocytes.17 ,15 However, the presence of mononuclear inflammatory cells, that is, macrophages and T lymphocytes, is not increased in VEGF tg mice of <3 months of age (see supplementary figure S29 ,16). Although mast cells might play a role,9 it seems that the proinflammatory effects of VEGF are primarily due to the induction of proinflammatory chemotactic cytokines including IL-8 and MCP-1 in microvascular endothelial cells,18 ,19 and TNFα, MIP-120 and IL-1321 in models of pulmonary fibrosis. Thus, the profibrotic effects partially depend on cytokines, which is further supported by the experiments in bleomycin-induced skin fibrosis in our study as well as by data from other experimental organ fibrosis models,22–24 and by gene transfer studies of VEGF165 in rabbit hindlimb skeletal muscles.25 The pathophysiology might be different in bleomycin-induced fibrosis. For example, in a murine model of pulmonary fibrosis, upon bleomycin challenge, there was an increase of VEGF levels, which was paralleled by augmented numbers of inflammatory cells, microvessels and cytokines levels, and was followed by tissue fibrosis. Abrogation of VEGF signalling at both an early and late fibrotic phase reduced the previously mentioned effects.23
However, VEGF also aggravated fibrosis under non-inflammatory conditions such as in TSK1 mice suggesting additional mechanisms. Accordingly, VEGF showed direct profibrotic effects on SSc fibroblasts by stimulating collagen production in our in vitro experiments. This direct profibrotic effect might be mediated via interaction of VEGF with platelet-derived growth factor receptors (PDGFRs) rather than VEGFRs, as SSc fibroblasts and lesional tissues express high amounts of PDGFR,26–29 but do not express VEGFRs, neither ex vivo8 nor in our current study in vitro (see supplementary figure S3). The PDGF class III receptor tyrosine kinase family30 is structurally related to the VEGFR family, and it has been shown that VEGF-A binds to PDGFRs inducing biologically functional signalling (proliferation/migration) in mesenchymal cells and fibroblasts.12 The increased collagen production in SSc compared with normal fibroblasts might be due to a persistent activation of PDGF signalling with an autocrine PDGF-A/PDGR-A loop with enhanced profibrotic responses.31 ,32
Alternative signalling pathways might also be involved. Although dermal fibroblasts do not express VEGFR, they express the VEGF coreceptors neuropilin-1 and neuropilin-2. These coreceptors might facilitate binding of VEGF-A to PDGFR, but are unlikely to independently mediate VEGF-dependent signalling effects. Additionally, VEGF-A is able to stimulate phosphorylation of epidermal growth factor receptor (EGFR) in mesenchymal cells.12 Enhanced activation of EGFR has been associated with the development and progression of fibrosis,33 possibly via induction of transforming growth factor beta receptor 2 (TGF-β-R2).34 Additionally, there is evidence that inhibition of VEGF signalling reduces TGF-β-mediated fibrosis.20 ,35
Furthermore, studies on scar formation suggest increased vascular permeability and the effects of an increased angiogenesis with secretion of profibrotic cytokines such as TGF-β or CTGF by endothelial cells as additional profibrotic mechanisms of VEGF.36
The lack of sufficient angiogenesis despite the strongly increased levels of VEGF and other proangiogenic factors remains one of the major enigmas in the pathogenesis of SSc. Recent data point to an important role of the inhibitory, antiangiogenic VEGF165b isoform that could at least partially explain this phenomenon.37 In addition, there are data suggesting that the chronic, uncontrolled and strong overexpression of VEGF as found in SSc might paradoxically lead to a chaotic capillary network with dilated capillaries rather than promoting the formation of new functional and stable capillaries.4 ,25 ,38 Our data in VEGF tg mice and the bleomycin skin fibrosis model support and add to this hypothesis by showing an inverse gene dosing effect with stronger angiogenesis in VEGF mono tg mice than in VEGF double tg mice. These data suggest that proangiogenic effects are best achieved with medium levels of VEGF, but not with high levels such as in VEGF double tg mice16 and in SSc patients.
In summary, our data on VEGF overexpressing mice and two different SSc models provide a novel molecular link between fibrosis and vasculopathy in the pathogenesis of SSc. They also show an inverse gene-dosing effect on the efficacy of angiogenesis. Thus, altogether these data suggest that high levels of VEGF potently induce fibrosis in inflammatory and non-inflammatory stages of SSc, and also contribute to the relatively insufficient angiogenesis characteristic for SSc.
Acknowledgments
We thank Michael Detmar for providing the VEGF tg mice and Maria Comazzi for the excellent technical support.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Handling editor Tore K Kvien
-
Contributors All authors have made a substantial contribution regarding the conception and design or analysis and interpretation of data. All authors have been involved in drafting the article or revising it critically for important intellectual content. All authors have given final approval of the presented version to be published.
-
Competing interests J.H.W. Distler has consultancy relationships and/or has received research funding from Boehringer Ingelheim, Celgene, Bayer Pharma, Actelion, Pfizer, Ergonex, BMS, JB Therapeutics, Anaphore Inc, Sanofi-Aventis, Novartis, Array Biopharma and Active Biotec in the area of potential treatments of scleroderma and is stock owner of 4D Science. O. Distler has/had consultancy relationship and/or has received research funding in the area of systemic sclerosis and related conditions from Actelion, Pfizer, Ergonex, BMS, Sanofi-Aventis, United BioSource Corporation, Roche/Genentech, medac, Biovitrium, Boehringer Ingelheim Pharma, Novartis, 4D Science, Active Biotec and Sinoxa. Yossra A. Suliman was supported by a Swiss Government grant. The other authors were supported by the respective institutions.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement Additional unpublished data from the study are available on request from the corresponding author.