Article Text

Abstract
Objectives Gouty arthritis patients for whom non-steroidal anti-inflammatory drugs and colchicine are inappropriate have limited treatment options. Canakinumab, an anti-interleukin-1β monoclonal antibody, may be an option for such patients. The authors assessed the efficacy/safety of one dose of canakinumab 150 mg (n=230) or triamcinolone acetonide (TA) 40 mg (n=226) at baseline and upon a new flare in frequently flaring patients contraindicated for, intolerant of, or unresponsive to non-steroidal anti-inflammatory drugs and/or colchicine. Core study co-primary endpoints were pain intensity 72 h postdose (0–100 mm visual analogue scale and time to first new flare.
Methods Two 12-week randomised, multicentre, active-controlled, double-blind, parallel-group core studies with double-blind 12-week extensions (response in acute flare and in prevention of episodes of re-flare in gout (β-RELIEVED and β-RELIEVED-II)).
Results 82.6% patients had comorbidities. Mean 72-h visual analogue scale pain score was lower with canakinumab (25.0 mm vs 35.7 mm; difference, −10.7 mm; 95% CI −15.4 to −6.0; p<0.0001), with significantly less physician-assessed tenderness and swelling (ORs=2.16 and 2.74; both p≤0.01) versus TA. Canakinumab significantly delayed time to first new flare, reduced the risk of new flares by 62% versus TA (HR: 0.38; 95% CI 0.26 to 0.57) in the core studies and by 56% (HR: 0.44; 95% CI 0.32 to 0.60; both p≤0.0001) over the entire 24-week period, and decreased median C-reactive protein levels (p≤0.0001 at 72 h and 7 days). Over the 24-week period, adverse events were reported in 66.2% (canakinumab) and 52.8% (TA) and serious adverse events were reported in 8.0% (canakinumab) and 3.5% (TA) of patients. Adverse events reported more frequently with canakinumab included infections, low neutrophil count and low platelet count.
Conclusion Canakinumab provided significant pain and inflammation relief and reduced the risk of new flares in these patients with acute gouty arthritis.
Statistics from Altmetric.com
Introduction
Gouty arthritis is the most common inflammatory arthritis in men,1,–,3 resulting from elevated body uric acid pools, which lead to deposition of monosodium urate crystals, mainly in the joints.4 ,5 Monosodium urate crystals can trigger the release of proinflammatory cytokines, in particular, interleukin-1β (IL-1β).3 ,6 Acutely, anti-inflammatory treatment with non-steroidal anti-inflammatory drugs (NSAIDs), colchicine, or corticosteroids can provide rapid pain relief and resolution of flares.7,–,9 Chronically, urate-lowering therapies (ULTs) reduce urate stores and, with anti-inflammatory prophylaxis, reduce the risk of new flares.8 ,9 Anti-inflammatory therapies may be inappropriate due to contraindications, intolerance, or unresponsiveness. Many of these patients have multiple comorbidities, such as hypertension, diabetes mellitus, renal insufficiency and cardiovascular disease,10 ,11 which can be worsened by these treatments.12 Consequently, these patients can develop frequent flares and persistent inflammation between flares,13 which may contribute to joint destruction and may impact health-related quality of life.14 ,15 In addition, uncontrolled gouty arthritis and hyperuricaemia may exacerbate comorbidities.16 ,17 Therefore, effective alternative treatments for flares are needed in this patient population.
As IL-1β is the key mediator in gouty arthritis inflammation, targeted anti-IL-1β therapy may be a valuable option for the management of gouty arthritis.18 Canakinumab, a fully human anti-IL-1β monoclonal antibody with a long plasma half-life (3–4 weeks)19 has shown superior efficacy (more rapid and sustained pain relief and significant reduction in risk of new flares) to triamcinolone acetonide (TA) in a dose-ranging study in patients with acute gouty arthritis.20 We report the results of two phase three randomised studies (response in acute flare and in prevention of episodes of re-flare in gout (β-RELIEVED and β-RELIEVED-II)) of canakinumab 150 mg for the treatment of acute flares in patients with gouty arthritis who had contraindications for, intolerance of, or unresponsiveness to NSAIDs and/or colchicine.
Methods
Study design
Both core studies were 12-week, multicentre, active-controlled, double-blind, parallel-group, double-dummy, phase three studies followed by a 12-week double-blind extension study. All studies were approved by a local independent ethics committee and performed in accordance with the ICH Harmonised Tripartite Guidelines on Good Clinical Practice and the ethical principles of the Declaration of Helsinki.
Patients
Patients were screened for eligibility at the time of, or before, an acute flare. The studies enrolled patients: aged 18–85 years, meeting the American College of Rheumatology 1977 preliminary criteria for the classification of acute arthritis of primary gout,21 with a history of ≥three self-reported flares in the previous 12 months, having an acute flare for ≤five days characterised by baseline pain intensity ≥50 mm on a 0–100 mm visual analogue scale (VAS), having contraindications for, intolerance of, or unresponsiveness to NSAIDs and/or colchicine (as determined by the investigator (see online supplementary text for additional information)), and with a body mass index (BMI) ≤45 kg/m2. Patients taking ULT were on a stable dose and regimen for at least 2 weeks prior to randomisation and were expected to remain on a stable regimen during the study. Key exclusion criteria included: use of specified pain relief medications or biologics (including corticosteroids, narcotics, paracetamol/acetaminophen, ibuprofen, colchicine, IL-blocker and tumour necrosis factor inhibitor) within specified periods prior to study entry (see online supplementary text for additional information), rheumatoid arthritis, infectious/septic arthritis, or other acute inflammatory arthritis, history of malignancy, active, chronic, or recurrent infections, including tuberculosis, or HIV infection or hepatitis B or C infection (see online supplementary text for additional information). Patients who completed the core studies were allowed to roll over into the extension studies (see online supplementary text for additional information).
Randomisation and masking
For the core studies, within 5 days of baseline flare onset, patients meeting inclusion/exclusion criteria were randomised (1:1) to receive canakinumab 150 mg by subcutaneous injection or TA 40 mg by intramuscular injection, both with placebo matching (see online supplementary text for additional information). All patients and investigators were blinded to the study treatment. For the extension studies, drug administration was based on the treatment assignment in the core studies. Visit 1 included screening, randomisation, assessment of baseline flare pain intensity and treatment. Patients experiencing a new flare visited the study site as soon as possible (within 5 days of flare onset) for treatment with the same baseline study drug (see online supplementary text). The minimum period between two consecutive study drug administrations was 14 days. Patients having difficulty tolerating their pain or experiencing a flare within 14 days of receiving the study medication could take rescue medication (see online supplementary text). During the extension studies, patients continued to be treated on-demand for any new flares.
Assessments
Patients recorded pain intensity in the most affected joint prior to treatment of each flare. Subsequent pain assessments were recorded in a diary at 6, 12, 24, 48 and 72 h and 4, 5, 6 and 7 days postdose using the 0–100 mm VAS. Efficacy and safety assessments were conducted at 24 and 72 h, 7 days, and 4, 8, 12, 16, 20 and 24 weeks after baseline, for retreatment of new flares, and at 3 and 7 days after retreatment. New flares were defined as a flare in a previously unaffected joint or a new flare in an affected joint where the previous flare had resolved completely. Patients recorded rescue medication use. Blood samples were assessed for inflammatory markers (C-reactive protein (CRP) and serum amyloid A (SAA)), haematology, chemistry (including serum urate), and immunogenicity (anticanakinumab antibodies),22 starting predose during baseline flare and continuing at subsequent visits. Adverse events (AEs) and serious AEs (SAEs) were reported throughout the core and extension studies.
The core studies had two co-primary efficacy endpoints: pain intensity (VAS score) in the most affected joint at 72 h postdose and time to first new flare over the first 12 weeks. Secondary outcomes included: time to first new flare over the entire 24 weeks, global assessments by patients and physicians (each evaluated on a 5-point Likert scale); assessment of joint tenderness (evaluated on 4-point scale), swelling (evaluated on a 3-point scale) and erythema (evaluated on a 3-point scale) by physicians; values of inflammatory markers; and safety and tolerability (see online supplementary text). The primary objectives of the extension studies were to evaluate the time to next flare and to determine the long-term safety profile of repeated on-demand use of canakinumab 150 mg subcutaneous in patients with gouty arthritis.
Sample size determination and statistical analyses
For the core studies, a sample size of 110 patients per group (per study) was considered to be adequate to produce an overall power for both co-primary endpoints of approximately 90%, based on the results of the phase 2 study.20 It was estimated that between 150 and 200 patients per study would enter the extension studies.
The primary objectives of the core studies were to demonstrate superiority of canakinumab 150 mg over TA 40 mg with respect to both co-primary endpoints; these were tested at a one-sided 2.5% level with no adjustment for multiplicity. Between-treatment differences in pain intensity in the target joint at 72 h postdose were assessed by an analysis of covariance, with treatment group, baseline VAS score, and BMI at baseline as covariates. Least-squares means and corresponding 95% CIs were determined. The time to first flare was analysed using Cox proportional hazard regression model, with treatment and BMI at baseline as explanatory variables. Kaplan–Meier estimates of the proportion of patients with first new gouty arthritis flare and associated 95% CIs calculated using Greenwood's formula are presented. The number of per-patient flares was analysed using a negative binomial generalised linear model (no adjustment for multiplicity) with BMI at baseline as an explanatory variable. Physician's assessment of joint tenderness and swelling, and physician and patient global assessments were made using proportional odds regression; erythema and rescue medication were analysed using logistic regression (see online supplementary text). No between group-group safety analyses were prespecified and no statistical comparisons of safety endpoints were planned or performed. In the extension studies, follow-up analyses were performed including all data from the core and extension studies.
For the core and extension studies, demographics, patient disposition and all efficacy endpoints were analysed using the full analysis set (ie, all patients as randomised in the core studies who received at least one dose of study drug, analysed according to assigned treatment), and all safety assessments were based on the safety set (ie, all patients who received study drug and had at least one postbaseline safety assessment, analysed according to received treatment).
Results
Patients
Of the 728 patients screened, 272 screening failures occurred, the majority of which were due to absence of a flare. Between December 2009 and June 2010, 456 patients were enrolled into two phase 3 studies; one in European and non-US countries (β-RELIEVED, N=230) and the other primarily in the US (β-RELIEVED-II, N=226; figure 1). Most patients completed the 12-week study periods (91.2%). Fewer patients discontinued canakinumab than TA (8.4% vs 9.2%, respectively; figure 1). The number of doses per patient by treatment is reported in the online supplementary table S1.

Patient disposition in β-RELIEVED (A) and β-RELIEVED-II (B).
Of the 230 patients who enrolled in the β-RELIEVED core study, 175 patients entered the extension study (canakinumab, n=90; TA, n=85). In both the core and the extension study, 7.8% (9/115) of patients in the canakinumab group and 13.0% (15/115) of patients in the TA group discontinued. The leading cause for discontinuation was loss to follow-up (4.3% (5/115)) in the canakinumab group and unsatisfactory therapeutic effect (4.3% (5/115)) in the TA group. Of 226 patients who enrolled in the β-RELIEVED-II core study, 160 patients entered the extension study (canakinumab, n=84; TA, n=76). In both the core and the extension study, 17.0% (19/112) of patients in the canakinumab group and 13.2% (15/114) of patients in the TA group discontinued, most commonly due to withdrawal of consent (8.9% and 6.1%, respectively) and loss to follow-up (5.4% and 2.6%, respectively).
Demographic and baseline disease characteristics were generally well balanced across treatment groups and between the core studies (table 1). Most (82.6%) patients had comorbidities, including hypertension (59.5%) and chronic kidney disease (CKD) stage 2–5 (83.7%; see online supplementary table S2). Details of contraindication, intolerance and unresponsiveness to standard treatments are provided in table 1. More patients were considered to be contraindicated for, intolerant of, or unresponsive to NSAIDs than to colchicine (91.0% vs 42.3%). There were no statistically significant differences between treatment groups for baseline demographics or disease characteristics. However, a greater proportion of patients in β-RELIEVED-II had monoarthritis, whereas a greater proportion of patients in β-RELIEVED had tophaceous disease and comorbidities, especially hypertension, obesity and CKD. ULT use was greater in β-RELIEVED than in β-RELIEVED-II (52.6% vs 31.9%) (see online supplementary table S3). Overall, 56% of patients received treatment within 2 days of the onset of the first flare.
Demographic and baseline characteristics (safety set)
Efficacy
In both studies, the two co-primary objectives were met.
Pain relief
At baseline, mean pain scores in the most affected joint were 74.1 mm. From 6 h postdose, mean pain scores were lower for canakinumab than for TA and the overall difference between groups was statistically significant from 24 h (from 12 h in β-RELIEVED and from 48 h in β-RELIEVED-II) (figure 2). At 72 h (primary endpoint), mean pain scores were lower for canakinumab (25.0 mm vs 35.7 mm) and the difference in pain score between treatments was −10.7 mm (95% CI −15.4 to −6.0; p<0.0001). Fewer canakinumab-treated patients took rescue medication (37.3% vs 54.6%; OR=0.47; p=0.0001), with oral corticosteroids being taken by 11.1% of patients receiving canakinumab versus 23.6% of patients receiving TA (see online supplementary table S4).
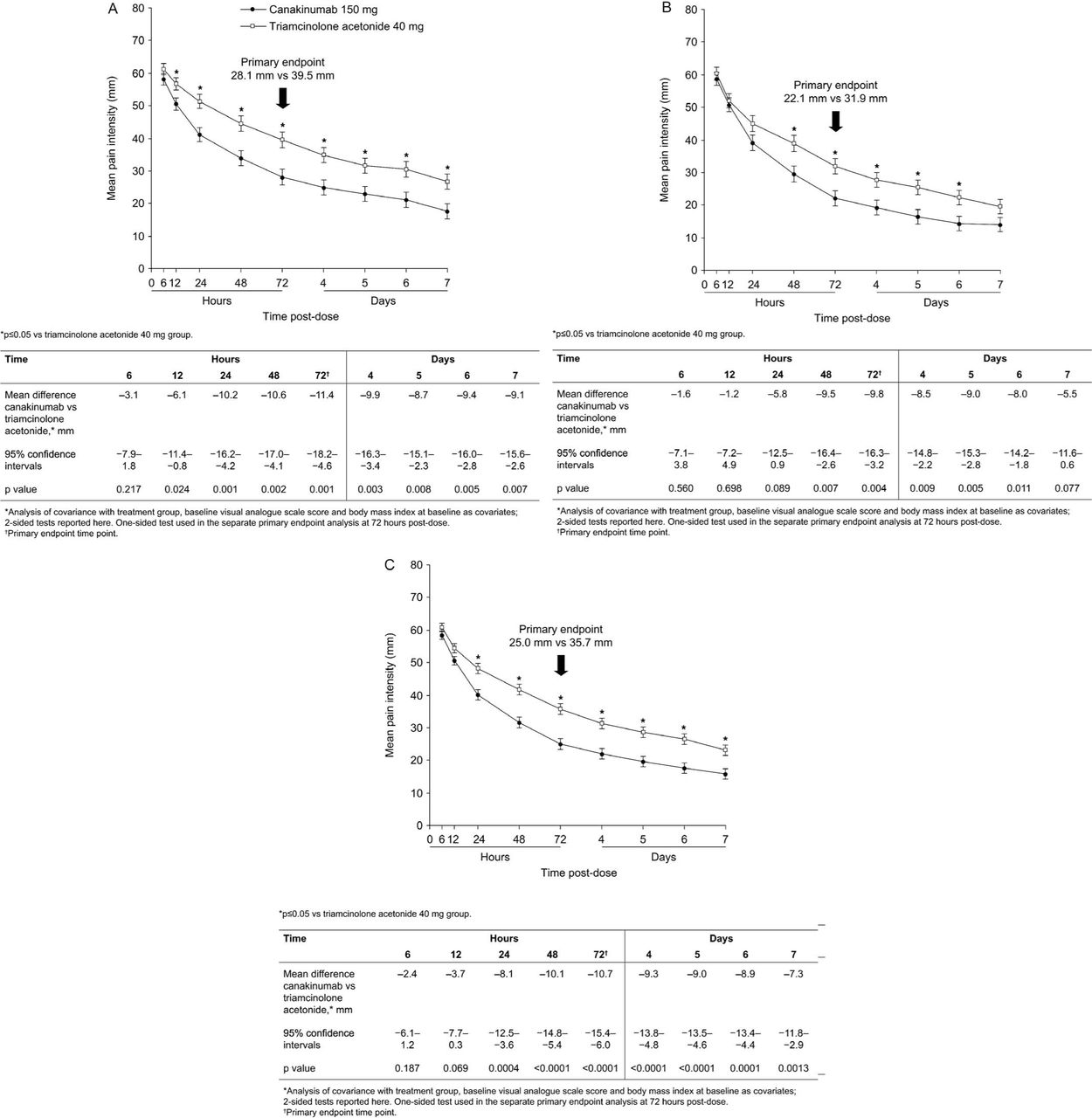
Least-squares mean pain intensity (0–100 mm visual analogue scale) over 7 days postdose for the first flare for β-RELIEVED (A), β-RELIEVED-II (B), and pooled data (C). Data are least-squares means ± #SEM
Reduction in the symptoms, signs and markers of inflammation
At 72 h, canakinumab was associated with significantly better responses to treatment versus TA according to patient global self-assessment (OR, 2.2; p≤0.0001) and physician global assessment (OR, 2.3; p≤0.0001), and significantly less tenderness (OR, 2.2; p≤0.0001), swelling (OR, 1.7; p≤0.01), and erythema (OR, 0.6; p≤0.05) (table 2; see also online supplementary table S5). Differences between treatments remained significant at 7 days postdose. Canakinumab produced rapid decreases in median levels of CRP and SAA; levels were consistently suppressed over the entire 24-week period and were lower at each time point in the canakinumab group (p≤0.0001 at 72 h and 7 days; table 2).
Response to therapy and reduction in signs of inflammation
Reduction in risk of new flare
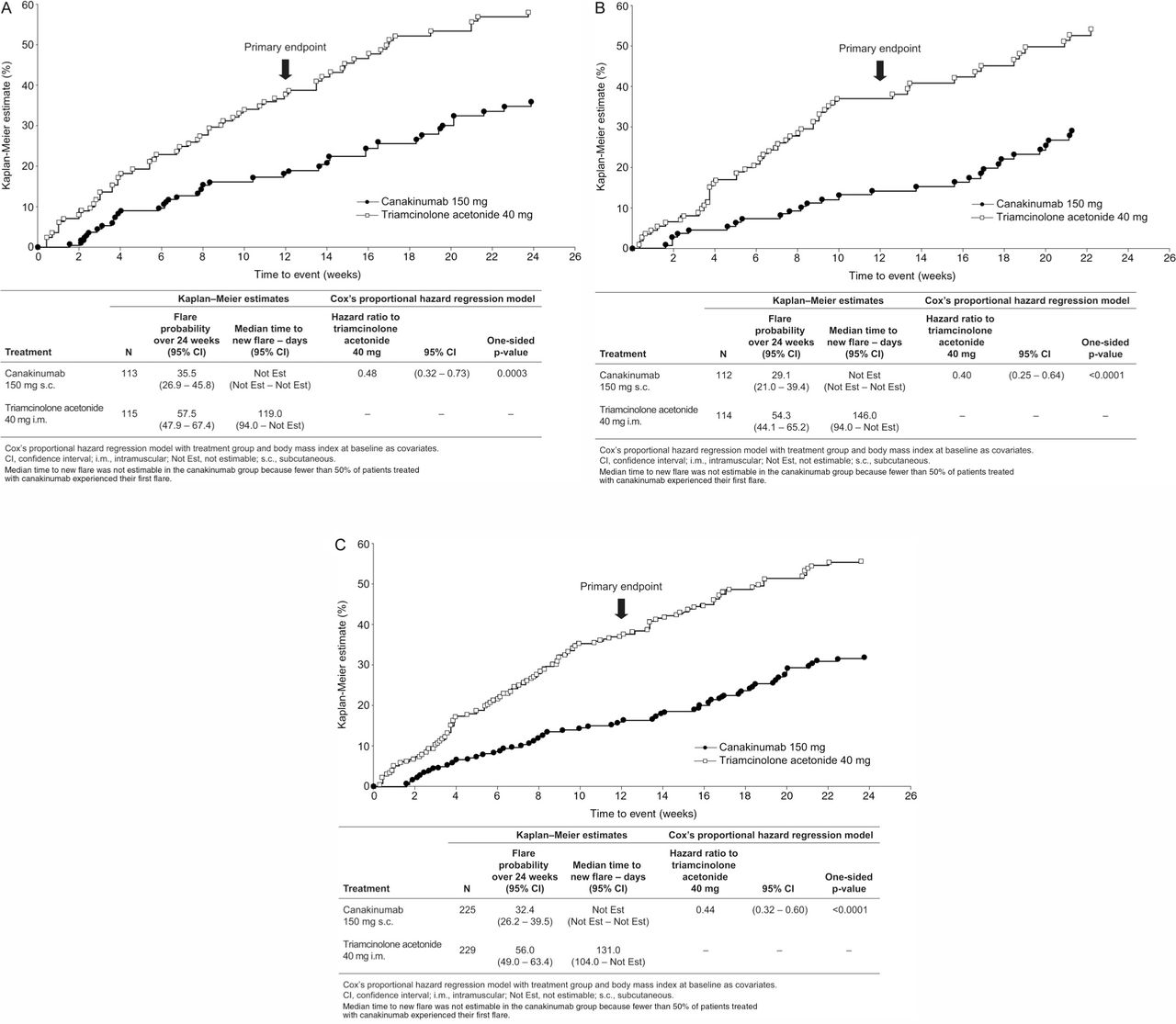
Canakinumab significantly delayed the time to first new flare and reduced the risk of a new flare over the 12-week period by 62% versus TA (p≤0.0001; figure 3). Fewer patients treated with canakinumab experienced at least one new flare during the core study (16.0% vs 35.8%; OR=0.34); the mean number of new flares per patient was significantly lower with canakinumab (0.19 vs 0.51; rate ratio=0.37; p<0.0001); and similarly, fewer canakinumab-treated patients had two or more new flares (2.7% vs 11.4%).

{kind=link}
{kind=link}
{kind=link}
Time to first new flare in β-RELIEVED (A), β-RELIEVED-II (B), and pooled data (C) (Kaplan–Meier estimates)
The effects of canakinumab were sustained throughout the extension studies, with a significant delay in time to first new flare and reduced risk of new flare over the 24-week period in both studies pooled by 56% compared with TA (HR 0.44; p≤0.0001; figure 3). Fewer canakinumab-treated patients had ≥2 new flares, and the mean number of per patient new flares was significantly lower with canakinumab (0.40 vs 0.87; p<0.0001). The median time to new flares was >168 days (study duration) with canakinumab compared with 131 days for TA (figure 3).
Safety
Adverse events were reported in 66.2% of canakinumab-treated patients and 52.8% of patients receiving TA over the 24 weeks of the core and extension studies (table 3); most were mild or moderate. Infections were reported in 20.4% (canakinumab) and 12.2% (TA) of patients, and serious infections (abscess jaw (right submandibular), abscess limb (left forearm), pneumonia (right acute) and gastroenteritis) in 1.8% and 0%, respectively. All four patients with serious infections during the 24-week study period were hospitalised, with three receiving antibiotics. All resolved with standard of care. No opportunistic infections were reported. One case (0.4%) of benign neoplasm (lipoma) was reported in patients receiving canakinumab.
Adverse events over 24 weeks (safety set)
The most frequently reported AEs are summarised in table 3. There were two deaths, neither considered by the investigators to be related to the study medication: one patient in the TA group (β-RELIEVED) died from a pulmonary embolism and one patient in the canakinumab group (β-RELIEVED-II) died from intracranial haemorrhage. Non-fatal SAEs were reported in 24 patients (canakinumab, n=17; TA, n=7; table 3). Apart from new flares, which required hospitalisation of two patients receiving TA, all SAEs in both treatment groups were single events not suspected to be related to study drug by the investigator. Further AE information is presented in the online supplementary tables S6 and S7.
Laboratory results are presented in full in the online supplementary tables S8–S11. These included an increase of serum urate of approximately 0.5 mg/dl compared with predose value during baseline flare in patients treated with canakinumab, changes in lipid parameters, and an increased incidence of predefined notable decreases in platelets, neutrophils and white blood cell counts for patients treated with canakinumab versus TA. Such notable haematological abnormalities were not associated with any serious infections or bleeding disorders. Only two patients (<1%) experienced injection-site reactions with canakinumab (one was mild and one was moderate). Based on the assay used to date, no patients treated with canakinumab developed anticanakinumab antibodies.
Discussion
Many patients with gouty arthritis experience frequent flares and have comorbidities that may limit their anti-inflammatory treatment options in an acute flare.12 For patients with contraindications to both NSAIDs and/or colchicine, treatment options are particularly limited, and there is an unmet medical need in this patient population. We demonstrated in these two phase 3 studies and their extensions that a single dose of canakinumab during an acute flare provides rapid and effective pain relief and prolonged suppression of flares and inflammation. Canakinumab was consistently superior to the active comparator TA and was generally well tolerated in this patient population with a high prevalence of multiple medical comorbidities. These phase 3 studies have longer study durations than the phase 2 study (24 weeks instead of 8) and more specifically defined inclusion criteria for contraindications and unresponsiveness to NSAIDs and/or colchicine. They are the only studies to date that specifically selected patients contraindicated for, intolerant of, or unresponsive to NSAIDs and/or colchicine. Most patients had long-standing severe disease, characterised by a high incidence of flares in the year before study entry, and a high prevalence of oligoarticular tophaceous disease and comorbidities, especially hypertension, obesity and CKD. Only 42% of patients were on ULT, suggesting suboptimal management of chronic gouty arthritis.
Canakinumab was associated with significantly less pain from 24 h postdose (p<0.01) despite greater rescue medication use, particularly oral corticosteroids, in the TA group. These findings confirm the results of a phase 2 dose-finding study of canakinumab in this setting.20
Two other anti-IL-1 agents, rilonacept and anakinra, have been investigated for treatment of acute and chronic gouty arthritis. In patients with chronic disease, rilonacept provided significant relief in a pilot study,23 but failed to improve pain relative to indomethacin in a randomised, controlled study in patients with acute gouty arthritis.24 Case cohort data suggest that daily treatment with anakinra can reduce pain in patients with gouty arthritis.25 ,26 Thus canakinumab is the only IL-1-targeted therapy that has consistently shown effective pain relief in three active-controlled studies. As reported here, TA 40 mg also provided clinically relevant pain relief at 72 h postdose, comparable with that reported in the literature for other corticosteroids.27
In our studies, a single dose of canakinumab was associated with a delayed time to a first new flare and a 56% reduction in the risk of a new flare versus TA over 24 weeks. The effect may not be limited to delaying the time and decreasing the risk of the first new flare but also to subsequent flares as shown by the lower incidence of patients with multiple flares with canakinumab compared with TA. As canakinumab may play an important role in controlling inflammation, it may provide a window of opportunity for initiation or acceleration of ULT. To this end, canakinumab has been shown to significantly reduce the risk of flares compared with colchicine in patients initiating ULT, thus providing further evidence for canakinumab's effect on reduction in risk of new flares and flare severity.28 Thus, the addition of prophylactic canakinumab to long-term ULT may facilitate use of an appropriate ULT dose and foster ULT compliance.
Canakinumab provided potent and durable suppression of inflammation, with reduced clinical signs of inflammation by 3 days (72 h) postdose. We report sustained suppression over 24 weeks of median CRP and SAA levels to below the upper limit of normal, consistent with the results of the phase 2 canakinumab study.20 These are the first studies to report the rapid normalisation of the inflammatory markers CRP and SAA in acute gouty arthritis, thus supporting evidence that IL-1β contributes to the regulation of CRP and SAA production.29 Evidence implicating IL-1β in bone erosion in patients with gouty arthritis suggests that potent suppression of IL-1β and the associated inflammation may slow joint destruction and hence disease progression.13
The following changes in laboratory parameters were noted during the extension studies: a modest increase in serum urate levels, decreased platelets, neutrophils and white cell counts and changes in lipids.
Canakinumab, similar to other anticytokines, is expected to increase the risk of infections, including serious infections.30,–,32 In the current studies, the rates of infection AEs and SAEs with canakinumab were increased over TA. All serious infections resolved with standard of care. Of note, no opportunistic infections were reported. A further potential risk of anticytokines is related to immunosuppression possibly leading to malignancies.30 In these studies, only one case of benign neoplasm (lipoma) has been reported in patients receiving canakinumab. However, larger patient numbers and much longer follow-up periods are needed to determine the risk for malignancies.33
Observed increase in serum urate levels with canakinumab use were modest and appeared to be partially transient. As there were no screening samples taken prior to baseline flare, we cannot determine whether, during baseline flare, the urate levels were decreased due to a possible uricosuric effect,34,–,37 with the increases seen after treatment of the flare reflecting a return to the preflare values. Therefore, there is still a degree of uncertainty around this observation, and any potential clinical relevance of this will need to be assessed further.
Over the entire 24-week period, most AEs were mild or moderate, and there were no deaths or SAEs reported as related to study medication, suggesting that canakinumab is a well-tolerated option for this patient population. Importantly, retreatment with canakinumab did not increase incidence of AEs or SAEs; however, these results should be interpreted with caution due to the small data set (n=60). Further, there were no other signals of specific organ toxicity, no patients treated with canakinumab developed anticanakinumab antibodies using the current available assay, and mild or moderate local reactions to canakinumab injections were reported in two patients (<1%). Additional follow-up of patients in these studies is underway to assess the long-term safety of canakinumab in patients who have gouty arthritis, particularly in patients who have received multiple canakinumab treatments.
The current studies had several limitations. First, not all patients were on ULT at baseline (42.3% on ULT); this was also reported in other studies,10 ,11 suggesting suboptimal therapy in many patients, reflecting the reality of the clinical setting where adherence to ULT is poor.38 ,39 Second, patients could be enrolled up to 5 days from the onset of flare, rather than 2 days as in some published studies.40 ,41 Hence, results may have been influenced by the self-limiting nature of the flare usually lasting 7 to 12 days. However, the proportion of patients who entered the study 4 or 5 days after onset of their flare was small (approximately 20%) and was similar across treatment groups in both studies; therefore, we do not believe that this had an impact on the outcome. Third, TA 40 mg may not be the optimal corticosteroid treatment for gouty arthritis given no evidence-based guidelines regarding corticosteroid therapy. However, we suggest that intramuscular TA 40 mg was an appropriate comparator for this study because it has an extended duration of action of up to several weeks42 and has the longest half-life of any intramuscular steroid, with no comparable long-acting agent currently available. In addition, the intramuscular route ensured full adherence to treatment and the efficacy observed in the phase 2 study20 and in these phase 3 studies is similar to published results for oral prednisolone.27 Importantly, there is no evidence that other doses of TA are more powerful than the 40-mg dose when treating acute gout flares, though individual patients might respond better to a higher dose. Future studies comparing canakinumab with other prophylaxis treatments may provide better comparisons of long-acting therapies. Another limitation of most of the published studies is the absence of crystal identification as an inclusion criteria; a limitation for all recent anti-inflammatory trials in acute gout, including etoricoxib,40 ,43 rilonacept,23 celecoxib,44 lumiracoxib45 and colchicine.46
A single dose of canakinumab effectively relieved pain and inflammation associated with acute gouty arthritis flares and effectively prolonged suppression of flares in patients contraindicated for, intolerant of, or unresponsive to NSAIDs and/or colchicine. Thus, canakinumab may be an important treatment option in a targeted population of patients with frequent gouty arthritis attacks who are unable to use NSAIDs and colchicine and in whom frequent use of corticosteroids is not considered appropriate.
Acknowledgments
The authors thank the patients and investigators who took part in this study. The authors take full responsibility for the content of the paper. The authors thank Dr Michael Shetzline and Dr Tamara Kiechle for their critical review of the manuscript, and Dr Peter Sallstig and Dr Udayasankar Arulmani (Novartis Pharma AG) for their contribution to the study design and critical review of the first manuscript drafts. They also thank Dr Kirstin Stricker (Medical Communication Leader, Novartis Pharma AG) for her contribution to the interpretation of the data, critical review of the paper, and for coordinating author discussions and their writing of the manuscript, and Dr Rowena Hughes, Dr Gemma Carter and Dr Fran Karo (Oxford PharmaGenesis) for medical writing support, editorial assistance, and collation and incorporation of comments from all authors. Such editorial help was funded by Novartis Pharma AG, Basel, Switzerland. They would also like to thank the following co-investigators: Dr Teo Franic, Dr Dick Quan and Dr Hsin Hua Liu (Holdsworth House Medical Practice, Sydney, Australia).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
-
Funding This study was supported by Novartis Pharma AG, Basel, Switzerland.
-
Competing interests NS reports having received a grant, travel expenses and payment for advisory board membership from Novartis Pharma, payment for advisory board membership and educational presentations from Takeda and Savient, and payment for advisory board membership from Savient, URL Pharma and Enzyme Rx. REA reports having received consulting fees from Novartis, payment for educational presentations from Novartis, Abbott and BMS, and payment for advisory board membership from Abbott, BMS and Roche. TB reports having received payments for advisory board membership, consultancy and educational presentations from Novartis, for consultancy from Savient, for advisory board membership and development of educational material from Ipsen and Menarini, and for development of educational material from Mayoli Spindler. MB reports having received travel expenses and payments for advisory board membership and educational presentations from Novartis. HRS reports having received grants and payments for consultancy and educational presentations from Takeda, grants and payments for consultancy from Pfizer, and payments for consultancy from Novartis, Regeneron, Savient, and Ardea Biosciences. DR, GK and VM are employees of Novartis and report having equity interests in Novartis. AG reports having equity interests in Novartis. AKS reports having received payment for board membership, consultancy and travel expenses from Novartis, and for educational presentations from Menarini.
-
Ethics approval Two global multicentre studies were involved (numerous Committees/IRBs reviewed).
-
Provenance and peer review Not commissioned; externally peer reviewed.