Article Text

Abstract
Objectives The P2X7 purinergic receptor antagonist AZD9056 was evaluated in a phase IIa study and subsequently in a phase IIb study to assess the effects of orally administered AZD9056 on the signs/symptoms of rheumatoid arthritis (RA), with American College of Rheumatology 20% response criteria (ACR20) as the primary outcome.
Methods Both studies were randomised, double-blind, placebo-controlled, parallel-group studies in patients with RA receiving methotrexate or sulphasalazine. Phase IIa was an ascending-dose trial in two cohorts (n=75) using AZD9056 administered daily over 4 weeks. Phase IIb included an open-label etanercept treatment group. Patients were randomised to receive treatment for 6 months with 50, 100, 200 or 400 mg AZD9056 (oral, once a day) or matching placebo (oral, once a day), or subcutaneous etanercept (50 mg once a week).
Results In phase IIa, 65% of AZD9056 recipients at 400 mg/day responded at the ACR20 level compared with 27% of placebo-treated patients. A significant reduction in swollen and tender joint count was observed in the actively treated group compared with placebo, whereas no effect on acute-phase response was observed. Of 385 randomised patients in the phase IIb study, 383 received treatment. AZD9056 (all doses) had no clinically or statistically significant effect on RA relative to placebo as measured by the proportion of patients meeting the ACR20 criteria at 6 months and further supported by secondary end points. In both studies AZD9056 was well tolerated up to 400 mg/day.
Conclusions AZD9056 does not have significant efficacy in the treatment of RA, and the P2X7 receptor does not appear to be a therapeutically useful target in RA.
Trial registration number ClinicalTrials.gov NCT00520572.
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is an immunologically mediated disease in which cytokines are key regulatory molecules. Interleukin (IL)-1β and IL-18 have been shown, in preclinical and clinical studies, to act synergistically in the pathogenesis of RA.1 ,2 IL-1, when targeted alone, has not delivered the magnitude of clinical benefit necessary for routine clinical use.3 ,4 Therefore, there is a rationale for simultaneously targeting IL-1β and IL-18 in the treatment of RA. P2X7 purinergic receptor is an ATP-gated ion channel involved in the processing and release of these cytokines. Recently, clinically useful ion channel blockers have been developed.5 ,6 The P2X7 receptor antagonist AZD9056 has been evaluated in preclinical models of RA with promising results (McInnes et al, unpublished data). We have evaluated AZD9056 in the clinical setting in a 1-month, randomised, placebo-controlled phase IIa clinical trial in RA patients and subsequently in a 6-month proof-of-concept phase IIb trial, which was designed to evaluate the dose–response relationship across four doses of AZD9056 (50, 100, 200 and 400 mg) on the signs and symptoms of RA.
Patients and methods
Clinical trial procedures – phase IIa
To formally investigate whether P2X7 mediates pro-inflammatory activity in the joint, we performed a phase IIa, dose-finding, placebo-controlled clinical trial (study ID: D1520C05287, CREATE study) using AZD9056. Ethics committees from each site approved the protocol and all patients gave written, informed consent before undergoing a screening evaluation to determine their eligibility.
Patients – phase IIa
Between July 2004 and April 2005, patients were screened for inclusion at seven centres in the UK and Romania. Eligible patients were those aged between 18 and 80 years who had active, adult-onset RA, defined as ≥6 swollen and ≥6 tender/painful joints plus either plasma C-reactive protein (CRP) ≥10 mg/l or erythrocyte sedimentation rate (ESR) ≥28 mm/h (following amendment of the entry criterion relating to CRP) and were American College of Rheumatology (ACR) functional class I–III. Patients had to have been treated with methotrexate and/or sulphasalazine for ≥12 weeks prior to randomisation, with no change to dose for the final 4 weeks. Patients treated with other disease-modifying agents were ineligible, as were patients who had received intramuscular or intra-articular steroid injection within 1 month of enrolment. Concurrent treatment with stable doses of non-steroidal anti-inflammatory drugs and/or prednisone (maximum 10 mg daily) was allowed throughout the study. Patients with current tuberculosis or any evidence of latent tuberculosis as determined by chest x-ray and skin test, or any other current infection or significant concurrent medical conditions, were excluded.
Study design
The study was a randomised, double-blind, placebo-controlled, parallel-group, ascending-dose trial in two cohorts. In total, 72 patients were required to be included in the study (36 per cohort). Patients were randomly assigned to receive either AZD9056 or placebo in a 2:1 ratio within each cohort, and also continued to receive stable doses of methotrexate and/or sulphasalazine. Randomisation was stratified by background therapy (methotrexate, sulphasalazine or both). AZD9056 and matching placebo were provided as tablets for oral administration. The study examined two doses of AZD9056: the higher dose of 400 mg for 28 days (cohort 2) was administered only after a comprehensive review of the safety of the first dose of 100 mg for 28 days (cohort 1).
The primary objectives of the study were to estimate tolerability of AZD9056 and to investigate the effects of P2X7 antagonism on signs and symptoms of RA, as measured by changes in composite measures of disease activity (ACR20 scores, Disease Activity Score 28 (DAS28)) and their individual components.
Statistical analysis of efficacy variables
At outset, the primary objective of the study was to assess safety and tolerability and the effect on CRP following drug exposure for 28 days. Patients entering the study initially required a significantly elevated CRP level of ≥15 mg/l. The primary objective and planned statistical analysis were modified in advance of unblinding of data. The amended primary objective was to investigate the clinical effects of AZD9056 on signs and symptoms of RA, as measured by changes in composite measures of disease activity (ACR scores, DAS28). Secondary objectives included evaluation of individual clinical response components, patient assessment of fatigue and morning stiffness, together with pharmacokinetic, pharmacodynamic and pharmacogenetic analyses. The efficacy analyses reported here were conducted using a per protocol analysis set defined a priori but were confirmed using the full analysis set (FAS). Placebo data from the two cohorts were pooled for the purpose of the analyses and a subsequent sensitivity analysis showed that this approach did not affect the overall conclusions. ACR20 response compared with placebo at 4 weeks was analysed using a χ2 test. DAS28 and Health Assessment Questionnaire Disability Index (HAQ-DI) were analysed using analysis of covariance for change from baseline at week 4, including treatment and baseline as a covariate. Median percentage changes from baseline in CRP and ESR compared with placebo after 4 weeks were analysed using the Wilcoxon rank sum test; non-parametric methods were used because the data were found to be right-skewed. p Values were calculated using the Mack–Skillings test. No formal statistical comparisons were performed between the two doses of AZD9056 for any variables. All other data were summarised using standard summary statistics. An independent interim efficacy analysis of ACR scores, DAS28 scores and their components for the 100 mg cohort showed that AZD9056 did not meet predefined efficacy criteria at this dose level. No adjustments were made for this in the subsequent analysis given the exploratory nature of the study.
Clinical trial procedures – phase IIb
The phase IIb study protocol (study ID: D1520C00001) was approved by an independent ethics committee/institutional review board, as appropriate, and was carried out in accordance with the Declaration of Helsinki and the International Conference on Harmonisation/Good Clinical Practice. All patients provided written informed consent.
Patients – phase IIb
Patients, ≥18 years of age, with diagnosed RA as defined by the ACR7 were eligible. Patients with active RA with an inadequate response to either methotrexate or sulphasalazine were evaluated. Active RA was defined as ≥4 swollen joints and ≥6 tender/painful joints (out of 28 joints), together with either ESR ≥28 mm/h or CRP ≥10 mg/l. Patients also had to have at least one of the following: documented history or current presence of positive rheumatoid factor; serum anticyclic citrullinated peptide (CCP) antibodies; baseline radiographic erosion. Patients were required to have received methotrexate for ≥6 months (the dose must have been stable between 5 and 25 mg/week for ≥6 weeks) or sulphasalazine for ≥16 weeks (at a stable dose of 0.5–3 g/day for ≥6 weeks) prior to randomisation.
Study design
The phase IIb study was a randomised, double-blind, placebo-controlled, parallel-group multicentre trial (with an open-label etanercept treatment group) to evaluate the efficacy of four doses of AZD9056 administered for 6 months on background methotrexate or sulphasalazine. Patients were randomly assigned (without formal stratification) in equal numbers (approximately 60 patients per group) to receive 6 months' treatment with 50, 100, 200 or 400 mg AZD9056 (oral, once daily), matching placebo (oral, once daily) or subcutaneous etanercept (50 mg once a week) as a positive control. Additional information on the statistical analyses is available in the online supplementary material.
The primary objective was to evaluate the proportion of patients meeting ACR 20% response criteria (ACR20) at 6 months (based on 28 joint counts). Secondary objectives included ACR50 and ACR70 criteria, changes in the individual ACR components, DAS28 remission rate (based on ESR), changes in DAS28, ESR, individual dimensions of the HAQ-DI, the patient's assessment of fatigue at 6 months, safety and tolerability of AZD9056, pharmacokinetics, quality of life (measured by the 36-item Short Form-36 (SF-36) and Rheumatoid Arthritis Quality of Life Questionnaires (RAQoL)) and radiological changes using standard x-ray (using the Sharp score as modified according to the method of van der Heijde).8
Results
Demographic and baseline characteristics
Phase IIa
In total, 75 RA patients were randomised to receive AZD9056 (100 mg/day, 400 mg/day) or placebo once daily for 4 weeks (see online supplementary figure 1). The 400 mg dose was administered following the satisfactory outcome of a formal safety review of patients receiving the 100 mg dose. Demographic analysis revealed balanced comparator cohorts (table 1).
Demographic and baseline characteristics for the phase IIa CREATE study

Clinical efficacy of AZD9056 in patients with rheumatoid arthritis from the phase IIa study. (A) ACR20 responses at week 2 and week 4. (B) Disease Activity Score 28 (DAS28) change from baseline. (C) Health Assessment Questionnaire Disability Index (HAQ-DI) change from baseline.
Phase IIb
A total of 385 patients were randomised, of whom 383 received treatment and were included in the FAS (online supplementary figure 2). Key baseline demographic and disease characteristics are shown in table 2. A total of 344 patients (89.8%) had previously received background therapy with methotrexate (median weekly dose 15 mg) and 37 patients (9.7%) had received sulphasalazine (median weekly dose 14 (range 3.5–28) g). Demographic and baseline characteristics were consistent with the expected study population. There were no notable differences between the treatment groups. Compliance with study medication was high: in each group more than 90% of patients were at least 80% compliant.

Clinical efficacy of AZD9056 in patients with rheumatoid arthritis from the phase IIb study. (A) Proportion of patients achieving American College of Rheumatology 20% response criteria (ACR20) at week 24. (B) ACR20 profile over time.
Efficacy results
Phase IIa
After 4 weeks, a significantly higher number of patients treated with AZD9056 400 mg (65%) achieved an ACR20 response compared with placebo recipients (27%; p<0.05; figure 1A). Consistent with this, the mean DAS28 improvement was 1.36 in the AZD9056 400 mg group compared with 0.89 in the placebo group (p<0.05; figure 1B). This was accounted for by a significant reduction in swollen and tender joint count that was observed in the AZD9056 400 mg group compared with placebo (p<0.05 and p<0.005, respectively), whereas no effect on acute-phase response was observed by week 4 (table 3).
Intermediate effects were observed in the 100 mg treatment group. There were no differences between groups with respect to ACR50 and ACR70 responses at 4 weeks. Finally, there was a significant, clinically meaningful improvement in function (measured by mean HAQ-DI) at 2 weeks in both the 100 mg (p<0.05) and 400 mg (p<0.01) groups (figure 1C).
Phase IIb
The proportion of patients achieving ACR20 at week 24 is shown in figure 2. The ACR20 responses of each of the AZD9056 treatment groups (50, 100, 200 and 400 mg) were similar to placebo. In contrast, the observed effect of etanercept was clearly distinguishable from placebo in all outcome measures. Extensive subgroup and sensitivity analyses supported the conclusions of the primary analysis (data not shown). The same pattern with AZD9056 was also observed with the other efficacy end points, the individual ACR components (online supplementary table 1), ACR50 and ACR70 criteria, ESR, DAS28, SF-36, RAQoL and fatigue, confirming the lack of clinically or statistically significant effect of AZD9056 in RA relative to placebo (data not shown). No systematic variation in response by baseline CRP level was observed, and no significant response was observed in any subgroup of patients with low, medium or high baseline CRP levels for any dose of AZD9056 (data not shown).
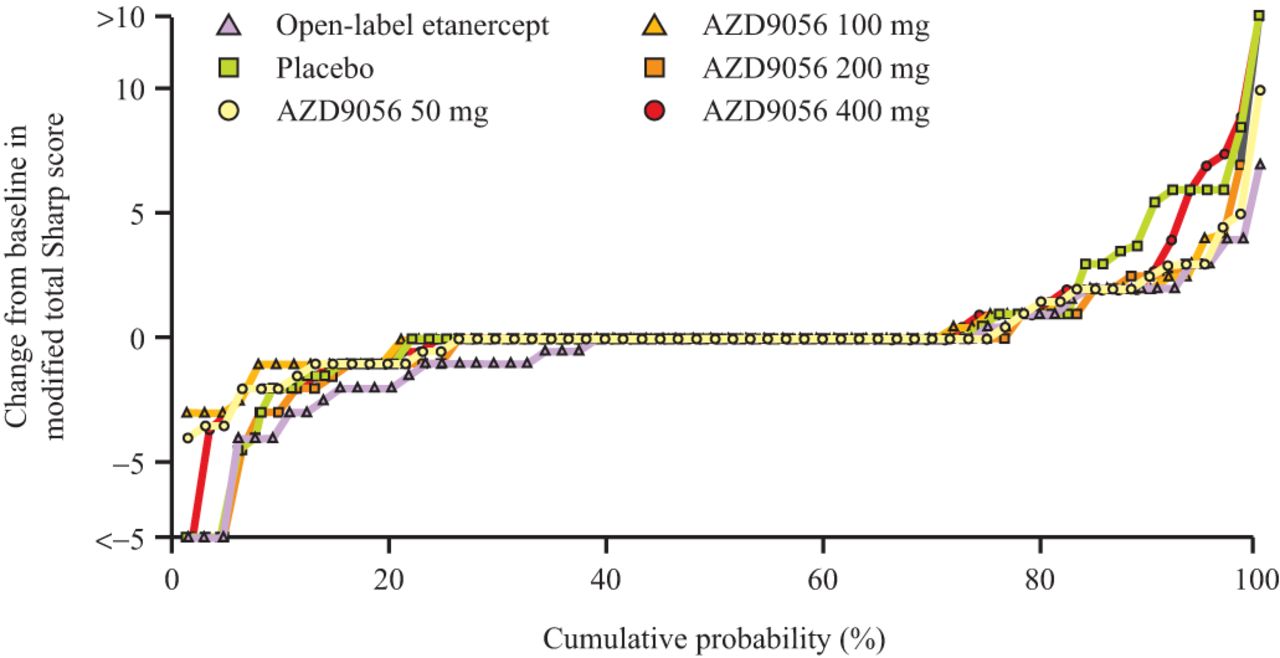
Radiographic progression was assessed by the cumulative probability plot of change in modified total Sharp score. The results raised the possibility of an effect of AZD9056 on radiographic progression (figure 3). Some separation from placebo was observed for the AZD9056 treatment groups in the total Sharp score (figure 3). This separation was apparent in the erosion score component, but not in the joint space narrowing. The observed effect in the open-label etanercept arm was clearly distinguishable from placebo.

{kind=link}
{kind=link}
{kind=link}
Structural progression, as measured by change from baseline in modified total Sharp score (cumulative probability), in the phase IIb study.
The percentage of rapid progressors (increase in Sharp score >5 by end of study) with AZD9056 at the lowest dose (1.7%) was similar to that observed with etanercept (1.6%), and lower than with placebo (11.1%). There appears to be an inverse dose effect in the percentage of rapid progressors, with the lowest AZD9056 dose similar to etanercept and the highest dose approaching placebo (online supplementary table 2). There was no significant difference between placebo and treatment arms in median change from baseline in total Sharp score.
Safety overview
AZD9056 was well tolerated in both dose cohorts of the phase IIa study. Nausea, vomiting and diarrhoea were reported more frequently following dosing with 400 mg AZD9056 than with 100 mg AZD9056 or placebo (see online supplementary table 3). Two 400 mg patients, one 100 mg patient and two placebo patients were withdrawn due to adverse events (see online supplementary table 4). There was one serious adverse event: a patient receiving AZD9056 400 mg was hospitalised after 3 days of treatment due to nausea and vomiting, which resolved the following day without discontinuation of AZD9056.
In the phase IIb trial, median exposure was comparable across the treatment groups (168 days in each of the AZD9056 arms, 169 days for placebo and 162 days for etanercept). AZD9056 was well tolerated up to 400 mg once daily. An overview of the incidence of adverse events is available in online supplementary table 5. There were no deaths due to adverse events and there were no serious infections reported. Seven patients (1.8%) experienced a serious adverse event, but this was considered by the investigator to be possibly related to treatment in only one case. The most common adverse events for AZD9056 by System Organ Class are listed in online supplementary table 6. Gastrointestinal adverse events were common, dose-related, and mainly mild to moderate in severity. No excess of infections or hepatic effects over placebo were reported. There was a small excess of visual and retinal adverse events in the AZD9056 arms. Vascular disorders occurring in patients in the AZD9056 and etanercept arms mainly consisted of hypertension.
Discussion
AZD9056, an adamantane amide, was discovered through a programme designed to identify potent and selective P2X7 antagonists. In a phase IIa study, AZD9056 (100 and 400 mg) once daily was generally well tolerated and induced statistically significant changes in parameters of clinical relevance (ACR20, DAS28, HAQ-DI). These responses were achieved despite no discernable change in acute-phase reactants (which comprise part of the composite outcome-measure set). Rather, the benefits were manifest mainly in reduced tender and swollen joint counts and in improved functional scores. In this respect, the human data parallel those seen in the streptococcal cell wall model (McInnes et al, unpublished data) and suggest that P2X7 antagonism, at least in the short term, could be acting primarily by altering local synovial processes without significantly altering systemic immune dysregulation manifest as elevated CRP or ESR. We also observed that AZD9056-treated healthy human volunteers exhibited significant inhibition of ex vivo ATP-induced monocyte IL-1 release (data not shown), suggesting that circulating leucocytes were blocked by P2X7. The phase IIa study with AZD9056 was encouraging and suggested a role for the P2X7 receptor as a therapeutic target in RA. These promising phase IIa data were, however, from a small study of short duration and required confirmation in a larger appropriately powered trial. The subsequent phase IIb study has shown that AZD9056 (50, 100, 200 and 400 mg) had no significantly greater efficacy than placebo in the treatment of the signs and symptoms of RA, as evaluated using ACR20 criteria. This was confirmed by consistent results with the ACR50 and ACR70 responses and all of the efficacy measures, including the individual ACR components and DAS28 scores. The validity of these data and the robustness of the study are supported by several important points. The characteristics of the current study population are similar to those in other comparable clinical trials in RA, having a sufficient baseline level of disease against which AZD9056 could be evaluated for efficacy. Administration of etanercept led to expected clinical responses, providing reassurance that the study population was amenable to improvement.9 Pharmacokinetic analysis showed that AZD9056 exposure was consistent with previous studies (data not shown) and that patients had the anticipated serum levels of drug. Furthermore, there were no significant findings on extensive subgroup and sensitivity analyses, performed to assess the impact of assumptions on the primary analysis conclusions and the robustness of the study. Only the radiographic data are suggestive of a possible effect on structural progression, although this should be treated with caution due to the small numbers of patients. The preclinical rationale for effects on bone was provided by a rat model of arthritis, in which antagonism of the P2X7 receptor attenuated joint destruction.10
The phase IIb study has demonstrated that data generated in ex vivo culture systems and in animal models as described by McInnes et al (unpublished data), which were suggestive of a positive effect of AZD9056 in RA, were not supported by favourable clinical outcomes. Moreover, the small phase IIa proof-of-concept study did not reflect the outcome in a larger, appropriately powered analysis. This has important implications for future trial design and in particular for the interpretation of early development programmes. First, we show once again that the interpretation of preclinical in vivo arthritis models requires caution, even in the context of plausible ex vivo biology.11 Several models may be required; notwithstanding this, their primary value should be in proof-of-pathway modulation (and corollary to relevant biological systems), rather than in establishing formal disease relevance. Second, we noted with interest the lack of robust, high-hurdle end points being achieved in the phase IIa study. In particular, no effect was noted on acute-phase reactants; clinical improvement lay largely in tender and swollen joint counts. These data emphasise the importance of relevant biomarker changes in early development studies. Finally, we conclude that the failure of IL-1 blockade in RA is not easily explained by functional redundancy in the IL-1 superfamily, or at least not by the activities of cytokines, the release of which is regulated by a P2X7 receptor-dependent mechanism, namely IL-1β and IL-18.
There is currently considerable interest in targeting ion channels from an immune regulatory perspective. They offer potentially attractive pharmacological opportunity in allowing the generation of small-molecule entities that in theory could mediate 'biological'-level responses. This is the first such study programme to be completed in humans, and the disappointing outcome will be informative in designing future approaches and selection of cell targeting.
Baseline demographics and disease activity for the phase IIb CREATE study
Change from baseline in key efficacy variables at weeks 2 and 4 for the phase IIa CREATE study
Acknowledgments
The authors would like to acknowledge all study investigators (listed in online supplementary material) and the following contributors: Carol Astbury and Noel Snell. The authors would like to thank Karen Brayshaw at Complete HealthVizion for medical writing assistance in the preparation and revision of the draft manuscript, based on detailed discussion and feedback from all the authors.
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
-
Funding This study was sponsored by AstraZeneca.
-
Competing interests EK has received funding for research from Abbott Laboratories; Amgen Inc.; AstraZeneca Pharmaceuticals LP; Bristol-Myers Squibb; Centocor, Inc.; F. Hoffmann-La Roche Inc.; Novartis Pharmaceuticals' Schering-Plough Corporation; UCB; and Wyeth Pharmaceuticals. EK had consulting agreements/advisory board membership with Abbott Laboratories; Amgen Inc.; Bristol-Myers Squibb Company; Centocor, Inc.; F. Hoffmann-La Roche Inc.; Genentech, Inc.; Schering-Plough Corporation; UCB; and Pfizer Pharmaceuticals. He also had speaker honoraria agreements with Abbott Laboratories; Amgen Inc.; Bristol-Myers Squibb Company; F. Hoffmann-La Roche Inc.; Schering-Plough Corporation; Pfizer Pharmaceuticals. MMW, ML and SH are employees of AstraZeneca and own stock. IBM has received grant funding and honoraria from AstraZeneca, F. Hoffmann-La Roche, Schering Plough, Bristol-Myers Squibb and Pfizer.
-
Patient consent Obtained.
-
Ethics approval Ethics committee approval obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.