Article Text

Abstract
Objective To investigate if statins prevent cartilage degradation and the production of collagenases and gelatinases in bovine nasal and human articular cartilage after proinflammatory cytokine stimulation.
Methods In a cartilage degradation model, the effects of several statins were assessed by measuring proteoglycan degradation and collagen degradation, while collagenolytic and gelatinolytic activity in culture supernatants were determined by collagen bioassay and gelatin zymography. The production of matrix metalloproteinases (MMPs) in cartilage and chondrocytes were analysed by real-time reverse transcriptase PCR and immunoassay. Cytokine-induced signalling pathway activation was studied by immunoblotting.
Results Simvastatin and mevastatin significantly inhibited interleukin 1 (IL-1)+oncostatin M (OSM)-induced collagen degradation; this was accompanied with a marked decrease in collagenase and gelatinase activity from bovine nasal cartilage. The cholesterol pathway intermediate mevalonic acid reversed the simvastatin-mediated protection of cartilage degradation, and the expression and production of collagenase (MMP-1 and MMP-13) and gelatinase (MMP-2 and MMP-9). Statins also significantly decreased MMP-1 and MMP-13 expression in human articular cartilage and chondrocytes stimulated with IL-1+OSM, and blocked the activation of critical proinflammatory signalling pathways required for MMP expression. The loss of the isoprenoid intermediate geranylgeranyl pyrophosphate due to statin treatment accounted for the inhibition of MMP expression and signalling pathway activation.
Conclusions This study shows, for the first time, that lipophilic statins are able to block cartilage collagen breakdown induced by proinflammatory cytokines, by downregulating key cartilage-degrading enzymes. This demonstrates a possible therapeutic role for statins in acting as anti-inflammatory agents and in protecting cartilage from damage in joint diseases.
Statistics from Altmetric.com
Introduction
Cartilage covers the articulating joint surface allowing free movement and is composed of an extracellular matrix (ECM) containing proteoglycan (aggrecan) and collagen (predominantly type II collagen). Irreversible progressive loss of the cartilage matrix is a major feature in rheumatoid arthritis (RA) and osteoarthritis (OA), which ultimately leads to the loss of joint function. Within this ECM chondrocytes control the turnover and remodelling of the cartilage matrix by regulating the expression of matrix components and matrix-degrading enzymes.1 Proinflammatory cytokines such as interleukin 1 (IL-1) and tumour necrosis factor α (TNFα) are considered as key pathogenic molecules in this process. Their action, especially when in combination with oncostatin M (OSM), a member of the IL-6 cytokine family, leads to an upregulation of a number of metalloproteinases (MPs), a family of zinc dependent endopeptidases that includes the matrix MPs and the ‘a disintegrin and metalloproteinase with thrombospondin motifs’ (ADAMTSs) which together have the ability to break down all ECM components.2 Aggrecan is degraded by MMPs and ADAMTSs, whereas type II collagen is degraded by MMPs, including MMPs 1, 8, 13 and 14. Following this cleavage gelatinases such as MMP-2 and MMP-9 and the stromelysin MMP-3 are able to further degrade collagen, while MMP-3 also has an important role in the activation of procollagenases and progelatinases.3 Together, these proteases release specific aggrecan or collagen II fragments that can be measured in vitro and in vivo.4 Loss of aggrecan from cartilage is reversible while collagen breakdown is not and therefore represents an irreversible step in cartilage degradation.5 6
Statins are the most commonly prescribed drugs for the prevention of cardiovascular diseases. They represent a group of 3-hydroxy-3-methylglutaryl (HMG) coenzyme A reductase inhibitors and thus reduce serum cholesterol levels by inhibiting the cholesterol biosynthesis pathway.7 Statin inhibition of HMG-coenzyme A reductase prevents the catalysation of HMG-coenzyme A to mevalonic acid (MA), the rate limiting step in the biosynthesis of cholesterol and the pathway intermediates such as the isoprenoids farnesyl pyrophosphate (FPP) and geranylgeranyl pyrophosphate (GGPP), which are involved in protein prenylation.8 Statins also possess anti-inflammatory properties and immunomodulatory actions that are unrelated to their lipid-lowering abilities and may be a result of the loss of protein prenylation.9 The isoprenoid intermediates are conjugated post-translationally to specific proteins containing a CAAX motif, in particular guanosine triphosphatases (GTPases) such as Ras, Rac and Rho family members, which have crucial roles in controlling many signalling pathways.10 11
As a result of these anti-inflammatory actions, statins have emerged as a class of drugs with potential for the treatment of rheumatic disorders, including RA. A small number of trials have attempted to address this, although only modest effects have been found so far and with qualifications.9 12 In animal models several studies have demonstrated that statins might inhibit the severity and progression of arthritis.13 15 In addition to their disease modifying activity, statins reduced the inflammatory cell infiltration into the synovium and downregulated proinflammatory cytokine levels.13 15 However, another study found no effect of a number of statins in a murine model of arthritis.16
We have previously reported that IL-1 or TNFα in combination with OSM synergistically induces cartilage degradation by upregulating MMPs, especially the collagenases MMP-1 and MMP-13, in vitro and in vivo.17 19 Statins have been studied in detail in the cardiovascular field and have been demonstrated to reduce MMP production.20 22 Statins can also block IL-1-induced MMP-3 in chondrocytes,23 and protein prenylation regulates IL-1 or TNFα-induced MMP-1 expression in rheumatoid synovial fibroblasts.24 However, no study to date has examined the ability of statins to prevent cartilage collagen breakdown, the critical pathological step in arthritic diseases. Therefore, in the present study, we investigate whether statins are able to directly protect cartilage against breakdown induced by proinflammatory cytokines, and if so, then to determine the mechanisms involved in the regulation of cartilage breakdown and chondrocyte MMP expression by statins.
Materials and methods
Materials
All cytokines used were recombinant human. IL-1α was a generous gift from Dr Keith Ray (Glaxo-SmithKline, Stevenage, UK). OSM was kindly donated by Professor John Heath (Department of Biochemistry, University of Birmingham, Edgbaston, UK). Simvastatin, mevastatin, fluvastatin and pravastatin sodium salts were purchased from Merck Chemicals (Nottingham, UK). MA, squalene, FPP and GGPP were from Sigma-Aldrich (Poole, UK). Primary antibodies used were phospho-extracellular signal-regulated kinase (ERK)1/2 (phospho-p44/42) (no. 9101), phospho-p38 (no. 9211), phospho-c-Jun N-terminal kinase (JNK) (no. 9251), phospho-Akt (Ser473; no. 9271) and nuclear factor κ light polypeptide gene enhancer in B cells inhibitor α (IκBα) (no. 9242) from Cell Signaling Technology (Danvers, Massachusetts, USA). A mouse monoclonal anti-glyceraldehyde 3′-phosphate dehydrogenase antibody (clone 6C5; MAB374) was purchased from Chemicon (Hampshire, UK). The polyclonal secondary immunoglobulins/horseradish peroxidase were from Cytomation (Dako, Glostrup, Denmark).
Cartilage assays
Bovine nasal septum cartilage and human articular cartilage were dissected into approximately 2×2×2-mm discs, plated into 24-well tissue culture plates (3 discs per well, n=4) and cultured in serum-free Dulbecco's modified Eagle medium as described previously.25 Fresh serum-free media with/without cytokines and test reagents were then added (day 0). At day 7, culture supernatants were harvested and replaced with fresh medium containing the same test reagents as day 0. Cartilage and culture supernatants were harvested at day 14 and the remaining cartilage was digested with papain. Hydroxyproline release was assayed as a measure of collagen degradation,26 and glycosaminoglycan release assayed as a measure of proteoglycan degradation.27 The extent of release was calculated as a percentage of the total. In a separate experiment culture supernatants were harvested at day 3 to assess early glycosaminoglycan release. Collagenase activities in the culture supernatants were determined by the 3H-acetylated collagen diffuse fibril assay with aminophenylmercuric acetate (0.67 mM) to activate procollagenases.28 Gelatinase activity in the culture supernatants was assayed by gelatin zymography.29 The culture supernatants were also assayed by specific immunoassay for MMP-130 and MMP-13.31
Cell culture
Human articular chondrocytes (HACs) were derived from articular cartilage obtained from consenting patients following hip or knee replacement surgery with Ethical Committee approval from the Newcastle and North Tyneside Health Authority, UK. Enzymatic digestion of tissue and maintenance and culture of cells were as previously described.17 When cells reached 80% to 90% confluence without passage they were pretreated with test reagents for the indicated timepoints after which cells were serum starved for 16 h prior to stimulation with cytokines for the indicated duration.
Cell viability
Viability of cartilage explants was assessed by screening for the production of lactate dehydrogenase (LDH) using the Cytotox 96 assay (Promega, Southampton, UK). Viability of HACs was assessed by screening for the production of adenylate kinase using the ToxiLight BioAssay Kit (Lonza, Rockland, Maine, USA).
RNA extraction from cartilage and chondrocytes
RNA was extracted from control and IL-1+OSM-stimulated cartilage at day 7 of the cartilage degradation assay. Cartilage was ground in a SPEX CertiPrep 6750 freezer mill (Glen Creston, Stanmore, UK) and total RNA was isolated from the powdered cartilage essentially as described29 with the use of TRIzol reagent (Invitrogen, Paisley, UK) and RNeasy Mini kit, including an on-column DNAse I digestion (Qiagen, Crawley, UK). cDNA was synthesised from 1 μg of total bovine cartilage RNA, using superscript II reverse transcriptase and random hexamers in a total volume of 20 μl according to the manufacturer's instructions (Invitrogen). From cultured HACs, total RNA was isolated and reverse transcribed using the ABI Cells-to-cDNA II Kit (Applied Biosystems, Foster City, California, USA).
Real-time reverse transcriptase PCR
Oligonucleotides and PCR Mastermix were purchased from Sigma-Genosys (Poole, UK), while TaqMan primer/probes were from Applied Biosystems as previously described.29 32 Relative quantitation of genes was performed using the ABI Prism 7900HT sequence detection system (Applied Biosystems). Bovine MMP expression profiles were determined using SYBR Green (Takara, Cambrex, Wokingham, UK) in accordance with the manufacturer's suggested protocol and as described.29 The 18S rRNA gene was used as an endogenous control to normalise for differences in the amount of total RNA present in each sample. Probe based MMP-1, MMP-13 and 18S assays were performed using TaqMan Mastermix reagents (Sigma-Aldrich) according to the manufacturer's protocol and as previously reported.32
Immunoblotting
Cells were lysed with ice-cold buffer (50 mM Tris-Cl, pH 7.5; 1.2 M glycerol; 1 mM ethylene glycol tetra-acetic acid (EGTA); 1 mM EDTA; 1 mM Na3VO4; 10 mM β-glycerophosphate; 50 mM NaF; 5 mM sodium pyrophosphate; 1% (v/v) Triton X-100; 1 μM microcystin-LR; 0.1% (v/v) β mercaptoethanol; Roche protease inhibitor complex), particulate matter removed by centrifugation at 13 000 g, 5 min at 4°C and lysates stored at –80°C until use. Lysates were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to polyvinylidene fluoride membranes (Millipore, Watford, UK) and subsequently probed using the antibodies described above. The quantitation of protein levels was performed with Adobe Photoshop CS (San Jose, California, USA) using a method available online (http://www.lukemiller.org/journal/2007/08/quantifying-western-blots-without.html).
Statistical analyses
Statistical differences between sample groups were assessed using the two-tailed Student t test, where *p<0.05; **p<0.01 and ***p<0.001. For quantification of immunoblots statistical differences between IL-1+OSM treatment and other treatment groups were assessed using the one-sample Student t test, where *p<0.05 and †p<0.01.
Results
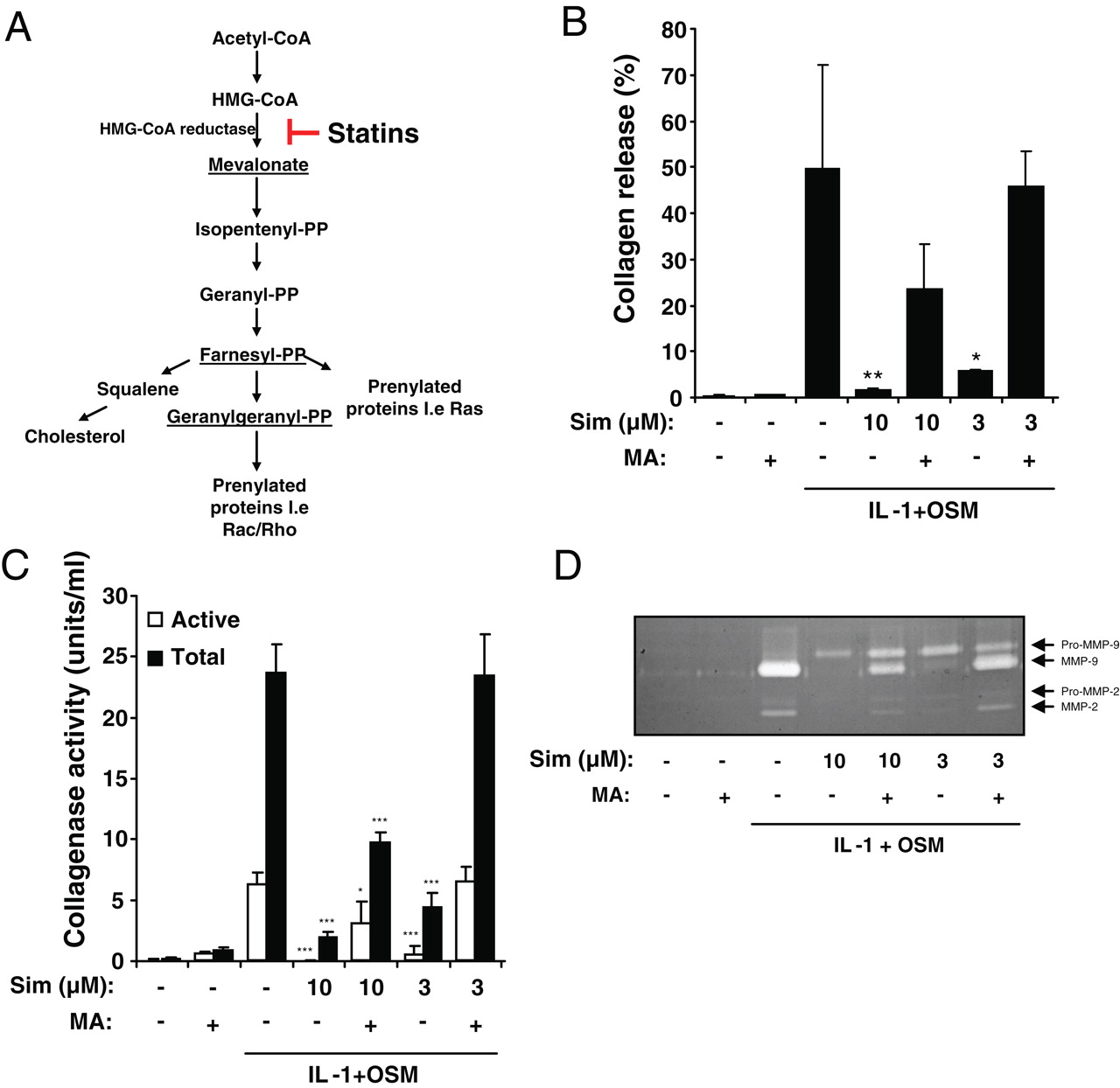
Effect of statins on the release of collagen from bovine nasal cartilage treated with IL-1+OSM
Previous studies have shown that IL-1 in combination with OSM can reproducibly stimulate the release of proteoglycan and collagen from bovine nasal cartilage in explant culture by day 7 and 14, respectively.17 18 33 Using this established model of cartilage destruction, we studied whether statins have a direct effect on cartilage degradation. Bovine nasal cartilage discs were cultured in control and IL-1α and OSM-containing media in the presence of mevastatin, simvastatin, fluvastatin, or pravastatin. After 14 days stimulation, IL-1+OSM induced over 80% collagen release from bovine nasal cartilage. Treatment with simvastatin or mevastatin significantly blocked IL-1+OSM-induced collagen release (figure 1A). Collagen release from TNFα+OSM treated cartilage was also similarly inhibited by simvastatin (data not shown). Fluvastatin also reduced collagen release, whereas the addition of pravastatin had no effect on the release of collagen. A concentration-dependent decrease in cartilage collagen degradation was observed with increasing concentrations of simvastatin and mevastatin (figure 1B). This was accompanied by reduced total and active collagenase activity (figure 1C). Statin treatment had no effect on the proteoglycan release from IL-1+OSM-stimulated bovine cartilage on day 7 (see supplementary material). Furthermore, simvastatin pretreatment for 3 days prior to IL-1+OSM treatment had no effect on day 3 proteoglycan release (see supplementary material). Neither mevastatin nor simvastatin affected the release of LDH from cartilage showing that, at the concentrations used, statins are not toxic to the chondrocytes (data not shown).

The effect of statins on proinflammatory cytokine-induced collagen release collagenolytic activity in bovine cartilage. Bovine nasal cartilage was cultured in serum-free medium in the presence of either medium alone, or medium containing interleukin 1α (IL-1α) (0.5 ng/ml) and oncostatin M (OSM) (10 ng/ml) with or without (A) the indicated statins at 10 µM, (B) simvastatin (Sim) or (B and C) mevastatin (Mev) at the indicated concentrations for 14 days. (A and B) The levels of collagen fragments released into the medium were determined by measurement of hydroxyproline as described in Materials and methods. Results shown are for the cumulative collagen release at day 14 of culture and expressed as a percentage of the total (mean±SD). (C) The levels of procollagenase and active collagenase activity in the media from cultured cartilage at day 14 were measured as described in Materials and methods (mean±SD). All assays were performed in quadruplicate. Significance was analysed with respect to IL-1+OSM with statin compared to IL-1+OSM alone, where *p<0.05, **p<0.01, ***p<0.001. Data are representative of three independent experiments.
Simvastatin and mevastatin prevent proinflammatory cytokine-induced collagenase production in human articular cartilage
To determine if statins are also able to protect human cartilage we measured the production of the collagenases, MMP-1 and MMP-13, following treatment of human articular cartilage explants with IL-1+OSM in the presence of a range of concentrations of simvastatin and mevastatin for 14 days. Levels of MMP-1 and MMP-13 in the culture supernatant were markedly elevated by IL-1+OSM and reduced by simvastatin and mevastatin in a concentration-dependent manner (figure 2A,B).

Statins prevent proinflammatory cytokine-induced matrix metalloproteinase (MMP) production in human articular cartilage. Human articular cartilage was cultured in serum-free medium in the presence of either medium alone, or medium containing interleukin 1 (IL-1) (0.5 ng/ml) and oncostatin M (OSM) (10 ng/ml) with or without the indicated concentrations of simvastatin (Sim) or mevastatin (Mev) for 14 days. The levels of (A) MMP-1 and (B) MMP-13 in human cartilage cultured medium were assayed by immunoassay as described in Materials and methods (mean±SD). All assays were performed in quadruplicate. Significance was analysed with respect to IL-1+OSM with statin compared to IL-1+OSM alone, where *p<0.05, **p<0.01, ***p<0.001. Data are representative of three independent experiments.
MA reversed the inhibition of cytokine-induced cartilage breakdown and MMP activity by simvastatin in bovine nasal cartilage
Focusing on the clinically relevant statin simvastatin, we sought to examine the mechanism by which statins prevent cartilage breakdown and block collagenase expression. Statins inhibit the enzyme HMG-coenzyme A reductase, preventing MA synthesis (figure 3A). MA is converted to cholesterol via the production of a number of intermediates including those required in protein prenylation, all of which are depleted following statin treatment. We studied the effect of addition of MA and the isoprenoids FPP and GGPP (underlined in figure 3A) in combination with simvastatin. MA fully reversed the effect of low concentrations of simvastatin (3 µM) on cartilage degradation and partially reversed the effect of the higher concentration of simvastatin (10 µM). Again, in collagenolytic assays using day 14 conditioned media from the cartilage degradation assay MA reversed the inhibitory effects of simvastatin (figure 3C) on IL-1+OSM-induced collagenase activity and expression. By gelatin zymography, IL-1+OSM resulted in the abundant release of active MMP-2 and MMP-9 into the conditioned media and this was significantly reduced following treatment with simvastatin and reversed upon addition of MA (figure 3D). These results indicated that the chondroprotection by statins is a result of the prevention of MA synthesis.

The effect of mevalonic acid (MA) on the inhibition of proinflammatory cytokine-induced collagen release by simvastatin from bovine cartilage. A. The MA pathway. Hydroxymethylglutaryl (HMG) coenzyme A reductase inhibitors (statins) block the conversion from HMG-coenzyme A to MA. Pathway intermediates used in this study are underlined. B–D. Bovine nasal cartilage was cultured in serum-free medium in the presence of either medium alone, or medium containing interleukin 1α (IL-1α) (0.5 ng/ml) and oncostatin M (OSM) (10 ng/ml) with or without simvastatin (3 or 10 μM) and the cholesterol biosynthesis pathway intermediate MA (100 µM) for 14 days. B. The levels of collagen fragments released into the medium were determined by measurement of hydroxyproline. Results shown are for the cumulative collagen release at day 14 of culture and expressed as a percentage of the total (mean±SD). C. The levels of procollagenase and active collagenase activity in the media from cultured cartilage at day 14 were measured (mean±SD). D. As a measure of gelatinase activity, the day 14 culture media were analysed by gelatin zymography as described in Materials and methods. All assays were performed in quadruplicate. Significance was analysed with respect to IL-1+OSM with statin compared to IL-1+OSM alone, where *p<0.05, ***p<0.001. Data are representative of three independent experiments.
MA reversed the effect of simvastatin on the inhibition of cytokine-induced MMP expression in bovine cartilage
We also investigated whether simvastatin blocked the expression of these key cartilage-degrading MMPs in bovine cartilage. Bovine cartilage was stimulated with IL-1+OSM in the presence of simvastatin ± MA for 7 days, after which total RNA was extracted and the expression of MMPs 1, 3, 9 and 13 assayed by reverse transcriptase PCR (RT-PCR). Simvastatin almost completely blocked IL-1+OSM-induced MMP-1 and MMP-13 expression and significantly decreased MMP-3 and MMP-9 expression (figure 4). Addition of MA partially reversed the downregulation of MMP-1, MMP-3 and MMP-13 by simvastatin, but had no effect on the expression of MMP-9. Since simvastatin did not prevent cytokine-mediated proteoglycan release we also determined the expression of the two major aggrecanases ADAMTS-4 and ADAMTS-5. Interestingly, simvastatin inhibited cytokine-induced ADAMTS-5 expression but had no significant effect on ADAMTS-4 (see supplementary material).

The effect of mevalonic acid (MA) on the inhibition of proinflammatory cytokine-induced matrix metalloproteinase (MMP) expression by simvastatin in bovine cartilage. Bovine nasal cartilage was cultured in serum-free medium in the presence of either medium alone, or medium containing interleukin 1α (IL-1α) (0.5 ng/ml) and oncostatin M (OSM) (10 ng/ml) with or without simvastatin (10 μM) and cholesterol biosynthesis pathway intermediate MA (100 μM) for 7 days. Total RNA was isolated as described in Materials and methods and subjected to real-time reverse transcriptase PCR for (A) MMP-1, (B) MMP-13, (C) MMP-3 and (D) MMP-9 mRNA expression normalised to 18S (n=3) (as indicated). Data are presented as fold induction relative to the basal expression and represent mean±SD. Significance was analysed with respect to IL-1+OSM with statin compared to IL-1+OSM alone, where *p<0.05. Data are representative of three independent experiments.
Loss of isoprenoid intermediates causes the simvastatin-mediated downregulation of MMP expression in cytokine stimulated human chondrocytes
To further determine the mechanism by which simvastatin blocked cartilage breakdown and MMP expression we assessed its effect in cultured primary HACs. HACs were pretreated with simvastatin for 0–72 h then stimulated with IL-1+OSM for 24 h and the expression of MMP-1 and MMP-13 was assayed by real-time RT-PCR. Pretreatment of HACs with simvastatin for 48 and 72 h significantly downregulated the expression of MMP-1 and MMP-13 induced by IL-1+OSM, whereas simvastatin pretreatment for 0 and 24 h had no effect (figure 5A). Cell viability, assessed by measuring adenylate kinase release, was unaffected by statin pretreatment (data not shown). Having established that 48 h statin pretreatment was required, HACs were then incubated with the cholesterol biosynthesis pathway intermediates in combination with simvastatin. Incubation with MA reversed the simvastatin-mediated inhibition of MMP expression (figure 5B). GGPP, and to a lesser extent FPP, also reversed the inhibition of MMP expression by simvastatin, suggesting that geranylgeranylation, and to a lesser extent farnesylation, of proteins is required for IL-1+OSM induction of MMP expression. In contrast, squalene had no effect on the simvastatin-mediated inhibition of MMP expression suggesting that this repression is not mediated by a loss of cholesterol synthesis. We also treated HACs with methyl-β-cyclodextrin, to remove cholesterol from the cell membrane,34 and found that this had no effect on the induction of MMP expression by IL-1+OSM (data not shown).

The effect of simvastatin treatment on proinflammatory cytokine-induced matrix metalloproteinase (MMP) expression in human chondrocytes. Human articular chondrocytes were stimulated with interleukin 1α (IL-1α) (0.02 ng/ml)+oncostatin M (OSM) (10 ng/ml) for 24 h with or without (A) pretreatment with simvastatin (10 μM) for the indicated times or (B) pretreatment with simvastatin (10 μM) for 48 h in the presence of the cholesterol biosynthesis pathway intermediates mevalonic acid (100 μM), farnesyl pyrophosphate (10 μM), geranylgeranyl pyrophosphate (10 μM) or squalene (50 μM). Total RNA was isolated and subjected to real-time reverse transcriptase PCR for MMP-1 and MMP-13 mRNA expression normalised to 18S (n=4). Data are presented as fold induction relative to the basal expression and represent mean±SD. Significance was analysed with respect to IL-1+OSM with statin compared to IL-1+OSM alone, where *p<0.05, **p<0.01, ***p<0.001. Data are representative of at least three independent experiments.
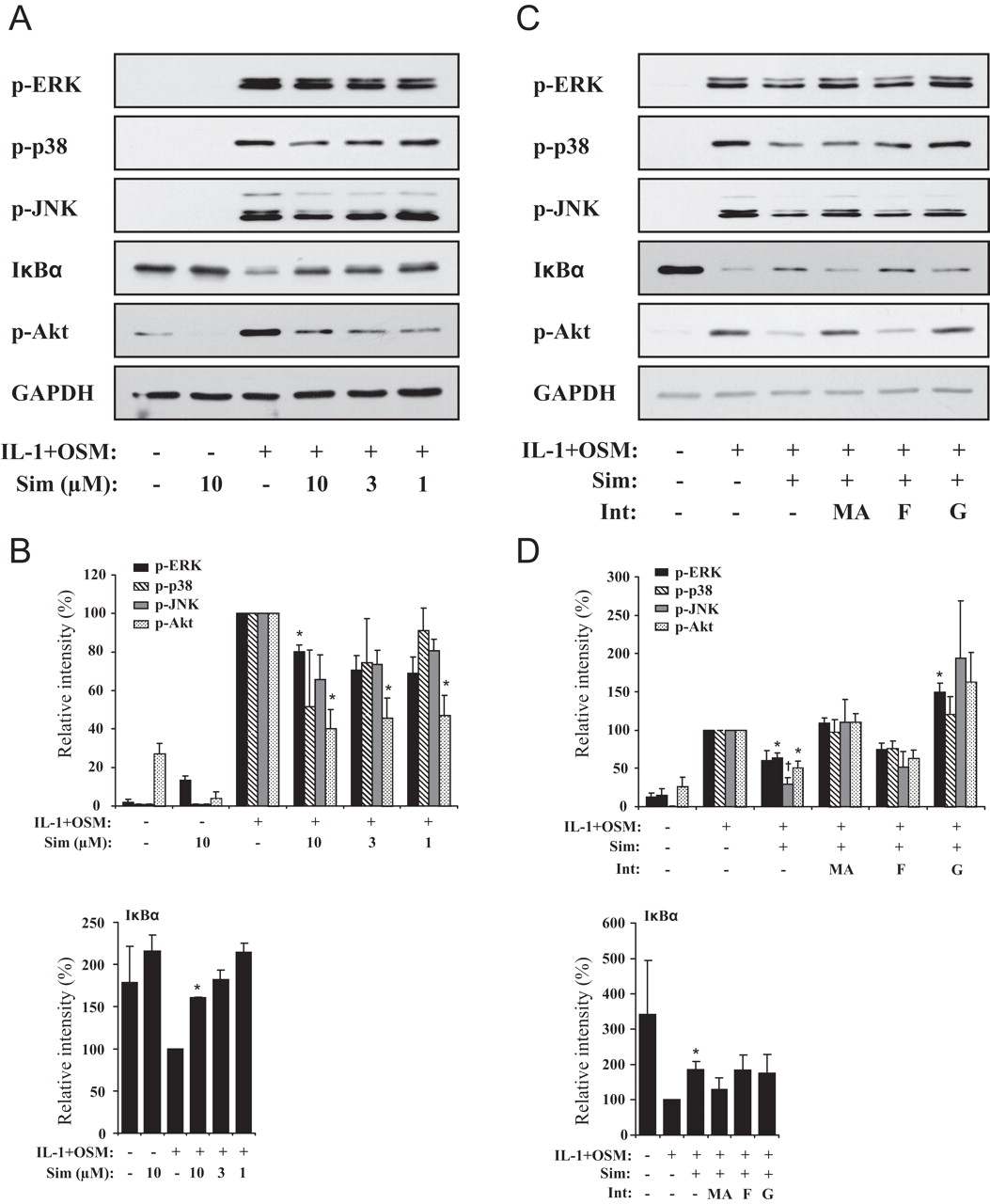
Effect of simvastatin on the cytokine-activation of signalling pathways in HACs
The proinflammatory cytokine induction of MMP expression in chondrocytes is mediated by the activation of a number of signalling pathways, in particular the mitogen-activated protein kinase (MAPK), nuclear factor (NF)κB and phosphoinositide 3-kinase (PI3K)/Akt pathways.35 36 The activation of these signalling pathways is regulated by small GTPases, which require prenylation by the pyrophosphate intermediates FPP and GGPP for their cell membrane-localised activity.10 11 Accordingly, we characterised the effect of simvastatin on the activation of signalling pathways by IL-1+OSM. Simvastatin treatment downregulated the activation of ERK, p38 and JNK in a concentration-dependent manner. Simvastatin also partially prevented IκBα degradation and significantly blocked the activation of Akt (figure 6A,B). To examine the mechanism we then incubated HACs with simvastatin in the presence of the cholesterol biosynthesis pathway intermediates. Consistent with its requirement in IL-1+OSM-induced cartilage degradation and MMP expression MA reversed the simvastatin-mediated inhibition of signalling pathway activation (figure 6C,D). GGPP also reversed the inhibition while FPP had no effect, suggesting that geranylgeranylation of proteins is necessary for the IL-1+OSM activation of signalling pathways in chondrocytes that mediate MMP expression.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The effect of simvastatin treatment on proinflammatory cytokine-induced signalling pathway activation in human chondrocytes. Human articular chondrocytes were stimulated with interleukin 1α (IL-1α) (0.02 ng/ml) and oncostatin M (OSM) (10 ng/ml) for 30 min with or without (A and B) pretreatment with simvastatin for 48 h at the indicated concentrations, or (C and D) pretreatment with simvastatin (10 μM) for 48 h in the presence of the cholesterol biosynthesis pathway intermediates mevalonic acid (100 μM), farnesyl pyrophosphate (10 μM) or geranylgeranyl pyrophosphate (10 μM). Total cellular protein was isolated as described in Materials and methods, separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis and immunoblotted with the indicated antibodies. Data are representative of at least three independent experiments. Quantification data are shown as mean±SEM percentage protein expression in comparison with IL-1+OSM alone levels for three independent experiments (A and B) and four independent experiments (C and D). Significance was analysed compared to IL-1+OSM alone, where *p<0.05 and †p<0.01.
Discussion
This study is the first to demonstrate that the lipophilic statins simvastatin and mevastatin directly protect cartilage from cytokine-mediated breakdown by preventing the upregulation in expression and production of collagen-degrading MMP-1 and MMP-13. This protective effect is mediated through the blocking of HMG-coenzyme A reductase MA generation and as a result reduced synthesis of the prenylation intermediate GGPP.
Statins, as widely used plasma cholesterol-lowering drugs have also been shown to have beneficial effects on a wide range of other conditions including atherosclerosis, nephropathy, bone disease, diabetes and cancer.37 Patients with RA have increased vascular risk and cardiovascular mortality and several clinical reports have suggested that statin treatment of patients with RA has a beneficial effect on disease activity, swollen joint count, endothelial dysfunction and arterial stiffness all linked with an improvement in markers of inflammation.38 In terms of OA, there are some epidemiological data supporting a link between OA and vascular disease,39 and OA is associated with mild inflammation,40 which taken together suggests that patients with OA may benefit from statin treatment.
Laboratory studies on the effect of statins in arthritis have focused on anti-inflammatory and immunomodulatory actions. Atorvastatin prevented joint inflammation in rat inflammatory arthritis with decreased tissue concentrations of IL-1β, IL-6, TNFα and chemokines.41 42 Statins also reduced the proportion of interferon γ (IFNγ) release by T cells and downregulated expression of inflammatory cytokines by synoviocytes in RA.13 15 In a murine collagen-induced arthritis model, simvastatin inhibited the progression of disease, even after the onset of arthritis and suppressed T helper (Th) 1 responses and IFNγ release.14 Mevastatin reduced cartilage degradation in a rabbit experimental OA model through inhibition of synovial inflammation with lowered expression of IL-1β, MMP-3, MMP-13 and monocyte chemoattractant protein 1 in synovial tissues, but not in cartilage.13 Statins can also induce RA synoviocytes to apoptose by the depletion of protein geranylgeranylation and loss of Ras homologue gene family, member A (RhoA) kinase activity.43 44
In this study we tested several clinically available statins and found that simvastatin, which accounts for approximately two-thirds of all UK statin prescriptions,45 and mevastatin, significantly prevented the proinflammatory cytokine IL-1+OSM-induced collagen degradation in bovine nasal cartilage. The actions of simvastatin and mevastatin were concentration dependent and were accompanied by a marked reduction in total and active collagenase and gelatinase activity. Mevastatin and simvastatin are lipophilic statins, which cross membranes non-selectively by passive diffusion, whereas pravastatin, a non-lipophilic statin, does not easily cross membranes.46 Fluvastatin has intermediate physicochemical properties and this was clearly reflected in our data with fluvastatin showing partial inhibition of cartilage degradation, whereas pravastatin had no effect. In contrast to collagen degradation statins were unable to prevent IL-1+OSM-induced proteoglycan degradation. It is possible that different mechanisms are involved in the activation of aggrecanase and collagenase activity, or that the cytokine combination used is more potent in inducing proteoglycan degradation than collagen. In murine arthritis ADAMTS-5 is reported to be the major aggrecanase.47 48 Therefore, it is perhaps surprising that simvastatin was able to inhibit ADAMTS-5 expression but not ADAMTS-4 without preventing proteoglycan release, especially when considering the prevailing dogma that ADAMTS-5 is the more catalytically active aggrecanase.49 One explanation could be that in bovine cartilage the absolute expression levels of both aggrecanases are considerably different, however, in our experiments the transcripts of ADAMTS-4 and ADAMTS-5 were of relatively equal abundance; although this similarity may not be reflected at the level of translation. It would also be important to determine the expression of inhibitors, especially TIMP-3, which is the more potent inhibitor of ADAMTSs,50 and how these are modified by statins. Finally, of course, the major aggrecanase in bovine nasal cartilage or human disease remains to be determined, with some evidence that ADAMTS-4 plays a role in human OA.51
Simvastatin significantly blocked MMP mRNA expression in bovine cartilage and statins also decreased the production of these MMPs in human OA articular cartilage and chondrocytes. This suggests that the chondroprotective effect of statins in cartilage is mainly mediated by downregulation of proteolytic enzymes, especially the collagenases. Throughout, the effects of statins were more profound on the cytokine-induced levels of MMP-13 as apposed to MMP-1. MMP-13 is more active against type II collagen than MMP-1,52 and is considered the primary collagenase driving degradation of cartilage collagen in OA.53 Of course, cartilage homeostasis is a balance between matrix synthesis and degradation, therefore, further studies are required to determine the effect of statins on the regulation of collagen and aggrecan gene expression. Any adverse effects in terms of matrix synthesis, coupled with an inability to protect against proteoglycan release, may explain the lack of clear efficacy of statins in OA trials (ref 54 and below).
Statins inhibit cholesterol synthesis at the level of the formation of MA and, as a consequence, also inhibit the production of various intermediates of the cholesterol biosynthesis pathway, including pyrophosphates required in the prenylation of GTPases.8 10 To investigate the cellular mechanism of statins on cartilage protection, we studied the role of these pathway intermediates in simvastatin mediated chondroprotection. MA reversed the effect of simvastatin on cartilage degradation, on simvastatin-inhibited active and total collagenase and gelatinase activity in cultured media, and on bovine and human MMP mRNA expression.
Previously, studies have found the activation of MAPK, NFκB and PI3K/Akt pathways by inflammatory cytokines such as IL-1, TNFα and OSM are highly involved in regulation of MMPs especially MMP-1 and MMP-13 expression in cartilage.32 35 36 In this study, using human OA articular chondrocytes to study the effect of statins on signalling pathways known to be regulated during an inflammatory response, we demonstrated that simvastatin treatment prevented the IL-1+OSM activation of MAPK, NFκB and PI3K/Akt pathways. Furthermore, we found that the inhibition of signalling pathway activation was mediated by the loss of protein geranylgeranylation, as addition of GGPP negated the effect of simvastatin. Cholesterol itself is also involved in cell signalling where it is found concentrated in lipid raft areas of the cell membrane in association with protein signalling complexes.55 However, addition of squalene was unable to reverse the inhibition of signalling pathways by simvastatin and the removal of cholesterol from the cell membrane by treatment with methyl-β-cyclodextrin had no effect on IL-1+OSM induced MMP expression.
It must be considered that the concentrations used to assess the effects of statins in the cartilage and chondrocyte cell cultures in this study are higher than clinically prescribed plasma concentrations which range between 0.1 and 1 µM for fluvastatin and 10-fold less for other statins.56 The statin concentrations used herein are identical to those routinely used to study their effect in in vitro cell studies.20 24 These concentrations may be necessary due to the greater activation of cells in vitro. In vivo the sustained lower plasma levels of statins might have a similar effect over time to those seen with the higher concentrations and shorter time periods used in vitro. Only a single trial has been published assessing statin use in OA and this actually associated the use of statins with increased risk of developing hip OA in women, although this could be due to other compounding factors. However, in the same study statins showed a trend toward a decreased risk of hip OA progression.54 The higher concentrations required to show efficacy in in vitro studies may explain why a significant effect has yet to be found in such trials. Accordingly, further studies are required to assess the impact of long-term statin administration in patients with arthritic diseases.
This is the first report to show that statins are able to directly prevent cytokine-induced cartilage collagen breakdown by reducing the expression and production of collagenases and gelatinases. MA in cartilage, and MA and GGPP in chondrocytes, reversed the effects of simvastatin indicating that the protective effect of simvastatin on cartilage is mainly through the prevention of protein geranylgeranylation. These findings suggest that statins can directly protect against cartilage damage and, given their well established anti-inflammatory and immunomodulatory role, have potential as therapeutic agents for inflammatory and non-inflammatory forms of arthritis.
Acknowledgments
This work was supported by Action Medical Research, the Newcastle upon Tyne Hospitals Special Trustees, the JGW Patterson Foundation, the Dunhill Medical Trust and the UK NIHR Biomedical Research Centre for Ageing and Age Related Disease Award to the Newcastle upon Tyne Foundation Hospitals NHS Trust.
References
Footnotes
-
Funding Action Medical Research.
-
Ethics approval This study was conducted with the approval of the Newcastle and North Tyneside Health Authority.
-
Provenance and peer review Not commissioned; externally peer reviewed.