Article Text

Abstract
Objective: To compare the efficacy, pharmacokinetics and safety of etanercept 50 mg once weekly with 25 mg twice weekly and placebo in patients with ankylosing spondylitis.
Methods: A 12-week, double-blind, placebo-controlled study compared the effects of etanercept 50 mg once weekly, etanercept 25 mg twice weekly and placebo in 356 patients with active ankylosing spondylitis (3:3:1 randomisation, respectively). The primary end point was the proportion of patients achieving a response at week 12 based on the Assessment in Ankylosing Spondylitis Working Group criteria (ASAS 20). The pharmacokinetics of etanercept 50 mg once weekly and 25 mg twice weekly were analysed.
Results: Baseline characteristics and disease activity were similar among the three groups: etanercept 50 mg once weekly, etanercept 25 mg twice weekly and placebo. The percentage of patients discontinuing therapy was 9.0%, 9.3% and 13.7% for the three respective groups. ASAS 20 response at 12 weeks was achieved by 74.2% of patients with etanercept 50 mg once weekly and 71.3% of those with etanercept 25 mg twice weekly, both significantly higher than the percentage of patients taking placebo (37.3%, p<0.001). Percentages of patients with ASAS 5/6 response (70.3%, 72.0% and 27.5%, respectively; p<0.001) and those with ASAS 40 response (58.1%, 53.3% and 21.6%, respectively; p<0.001) followed a similar pattern. Significant improvement (p<0.05) was seen in measures of disease activity, back pain, morning stiffness and C reactive protein levels as early as 2 weeks. Serum etanercept exposure was similar between the etanercept groups. Incidence of treatment-emergent adverse events, including infections, was similar among all three groups, and no unexpected safety issues were identified.
Conclusions: Patients with ankylosing spondylitis can expect a comparable significant improvement in clinical outcomes with similar safety when treated with etanercept 50 mg once weekly or with 25 mg twice weekly.
- ASAS, Assessment in Ankylosing Spondylitis
- AUC, area under the curve
- BASDAI, Bath Ankylosing Spondylitis Disease Activities Index
- CRP, C reactive protein
- NCI, National Cancer Institute
- TNF, tumour necrosis factor
- VAS, visual analogue scale
Statistics from Altmetric.com
- ASAS, Assessment in Ankylosing Spondylitis
- AUC, area under the curve
- BASDAI, Bath Ankylosing Spondylitis Disease Activities Index
- CRP, C reactive protein
- NCI, National Cancer Institute
- TNF, tumour necrosis factor
- VAS, visual analogue scale
Tumour necrosis factor (TNF)α is an important mediator of inflammation in rheumatic diseases such as ankylosing spondylitis, rheumatoid arthritis and psoriatic arthritis. Inhibition of TNFα reduces arthritic symptoms and joint injury in animal models of rheumatic disease.1 Etanercept, a recombinant human TNFα receptor, binds to TNFα and inhibits its activity.2 In clinical trials, etanercept at a dosage of 25 mg twice weekly is effective in the treatment of rheumatoid arthritis,3–7 ankylosing spondylitis,8,9,10,11,12,13 psoriatic arthritis14 and psoriasis,15,16 and has a well-defined and acceptable safety profile.
In patients with rheumatoid arthritis, the efficacy, safety and pharmacokinetics of etanercept 50 mg once weekly are comparable with those of etanercept 25 mg twice weekly.17 In North America and Europe, both dosages, are approved for the treatment of rheumatoid arthritis. As disease state can affect pharmacokinetics, the results observed in rheumatoid arthritis could hypothetically be different from those observed in ankylosing spondylitis. Therefore, the question of whether administration of etanercept once weekly is comparable with etanercept twice weekly requires assessment in patients with ankylosing spondylitis.
A double-blind, placebo-controlled study was conducted, evaluating the more convenient 50 mg-once-weekly regimen in patients with active ankylosing spondylitis. We report the results of this study comparing the efficacy, safety and pharmacokinetics of etanercept 50 mg once weekly, etanercept 25 mg twice weekly and placebo in patients with ankylosing spondylitis.
PATIENTS AND METHODS
Study design
The trial was a 12-week, randomised, double-blind, placebo-controlled, multicentre study with three treatment groups in a 3:3:1 ratio—etanercept 50 mg once weekly, etanercept 25 mg twice weekly and placebo, respectively. It was designed to determine the non-inferiority of etanercept 50 mg once weekly to etanercept 25 mg twice weekly with respect to Assessment in Ankylosing Spondylitis Working Group criteria (ASAS 20) at week 12. The placebo group provided a valid comparison to quantify the magnitude of the treatment effect. Patients were evaluated at screening, baseline and at weeks 2, 4, 8 and 12.
This study, which was carried out at 38 centres in 11 countries in Europe, including Belgium, France, Germany, Greece, Hungary, Italy, The Netherlands, Poland, Portugal, Spain and the UK, was conducted in accordance with the ethical principles of the Declaration of Helsinki and was consistent with the guidelines for good clinical practice. The study protocol and informed consent document were approved by each institution’s review board or independent ethics committee.
Patients
The study enrolled adult patients, aged 18–70 years, with active ankylosing spondylitis based on the Modified New York Criteria for ankylosing spondylitis.18 Active ankylosing spondylitis was defined by an average visual analogue scale (VAS) score ⩾30 for duration and intensity of morning stiffness and two or more of the following: patient global assessment of disease activity VAS score ⩾30; mean of nocturnal and total pain VAS scores ⩾30; or Bath Ankylosing Spondylitis Functional Index ⩾30 (all scores on a scale of 0–100). Concomitant oral non-steroidal anti-inflammatory drugs and oral corticosteroids (⩽10 mg/day), if stable for ⩾2 weeks before randomisation, and disease-modifying antirheumatic drugs (hydroxychloroquine, sulfasalazine and methotrexate), if stable for ⩾4 weeks before randomisation, were permitted.
Patients previously treated with TNFα inhibitors, including etanercept or other biological agents, or disease-modifying antirheumatic drugs (other than hydrochloroquine, sulfasalazine and methotrexate) less than 4 weeks before baseline, were not eligible. Other important exclusion criteria included complete ankylosis (fusion) of the spine based on radiographic assessment and concurrent medical events, such as uncontrolled hypertension, unstable angina pectoris, congestive heart failure, severe pulmonary disease, cancer, demyelinating diseases of the central nervous system and serious infections.
Assessments
The primary efficacy end point was the proportion of patients who achieved a response according to the ASAS 20 response criteria19 at week 12. An ASAS 20 response is fulfilled if there is at least a 20% improvement with a minimum of 10 units in at least three of four domains (pain, function, inflammation and patient global assessment), without worsening in the possible fourth domain by 20% and 10 units.
Secondary end points included the proportion of responders based on ASAS 4020 and ASAS 5/620 criteria at all time points. ASAS 40 is based on the same domains as ASAS 20, but requires at least a 40% improvement and 20 units in at least three of the four domains and no worsening in the remaining domain.20 ASAS 5/6 responders are defined as patients showing ⩾20% improvement in five of six domains: the four domains in ASAS 20 and C reactive protein (CRP) levels, and spinal mobility (modified Schober’s test).20
Other secondary end points included patient and physician global assessments of disease activity, nocturnal and total back pain assessments, Bath Ankylosing Spondylitis Functional Index,21 Bath Ankylosing Spondylitis Disease Activities Index (BASDAI),22 patients achieving partial remission, time to partial remission,19 spinal mobility (modified Schober’s test, chest expansion measurement and occiput-to-wall distance), joint assessment (70 joints) and serum CRP.
The pharmacokinetics of etanercept 50 mg once weekly and 25 mg twice weekly were analysed from blood samples collected at baseline and at weeks 2, 4 and 12, or at early withdrawal. Serum etanercept concentrations were determined using an enzyme-linked immunosorbent assay.
Safety assessments were based on reports of adverse events, routine physical examinations and laboratory test results. Treatment-emergent adverse events were defined as adverse events not present at baseline or, if present at baseline, worsened during the study. Medically important infections required hospitalisation or parenteral antimicrobial agents.
Statistical methods
The modified intent-to-treat population was the primary population for efficacy and safety analyses, and comprised all patients who received at least one dose of the test drugs. Demographic and baseline characteristics were analysed using one-way analysis of variance for continuous variables and Fisher’s exact or the χ2 test for categorical variables.
The study was designed to test the non-inferiority of etanercept 50 mg once weekly to 25 mg twice weekly at week 12. Non-inferiority between the two doses could be claimed if the lower bound of the one-sided 97.5% confidence interval (CI) of their differences in the primary end point at week 12 was above the non-inferiority margin of −18.5%. This −18.5% margin represented 50% preservation of the active treatment group effect (25 mg twice weekly) observed in a previous study,8 in which the ASAS 20 response rates were 60% and 23% in the etanercept and placebo groups, respectively.
For the secondary analyses, two-sided Fisher’s exact tests were conducted. A last-observation-carried-forward approach was used to impute missing data in the modified intent-to-treat population analysis.
RESULTS
Patient characteristics and disposition
Of the 361 patients, 356 received at least one dose of test drug and they comprised the modified intent-to-treat population. Demographic and other characteristics were not significantly different among the groups at baseline, except for history of inflammatory bowel disease (table 1).
Demographic and other baseline characteristics of the modified intent-to-treat population
A total of 35 (9.8%) patients discontinued before week 12 (table 2). Adverse events and lack of efficacy were the most common reasons for discontinuation in patients receiving etanercept and placebo, respectively; however, these differences were not significant among treatments (p<0.276 and p<0.184, respectively).
Patients who withdrew from the study by primary reason
Clinical efficacy
The primary end point, the ASAS 20 response rate at week 12, was achieved by significantly more patients receiving etanercept 50 mg once weekly (74.2%) or 25 mg twice weekly (71.3%) than those receiving placebo (37.3%; p<0).
Non-inferiority of etanercept 50 mg once weekly to 25 mg twice weekly was proved because the lower limit of the CI for the actual response proportion difference between treatment groups was −7.1%, which was greater than the protocol-defined non-inferiority margin of −18.5% (fig 1). In addition, the two-sided 95% CI (−7.1% to 12.9%) contained zero, confirming no significant difference between the two etanercept groups. A retrospective analysis, based on preserving at least 75% of the treatment effect, showed that the lower limit of the 95% CI was still greater than the non-inferiority margin (−9.25%). Therefore, even when a more stringent definition is applied, the etanercept 50 mg once weekly regimen remains non-inferior to the 25 mg twice weekly regimen.

Patients (%) achieving an Assessment in Ankylosing Spondylitis 20 response at week 12 in the etanercept-treatment groups and the demonstration of non-inferiority of etanercept 50 mg once weekly (QW) to etanercept 25 mg twice weekly (BIW; modified intent-to-treat population; last-observation-carried-forward approach).
Figure 2 shows the ASAS 20, ASAS 40 and ASAS 5/6 responses over the duration of the study. For all of the ASAS response criteria, the proportion of patients in either etanercept treatment group showing a response was significantly greater than that in the placebo-treated group. At week 12, almost three of every four patients receiving etanercept (70.3% and 72.0%, respectively, from the 50 mg-once-weekly and 25 mg-twice-weekly groups) achieved the ASAS 5/6 response compared with 27.5% of patients from the placebo group (p<0.001). Similar response rates were observed using the ASAS 40 cut-offs: 58.1% and 53.3%, respectively, for the etanercept 50 mg-once-weekly and 25 mg-twice-weekly groups versus 21.6% for the placebo group (p<0.001). Proportions of patients achieving the ASAS responses were not significantly different between the two etanercept groups at any time point.

Assessment in Ankylosing Spondylitis responders (%) over time (modified intent-to-treat population; last-observation-carried-forward approach). (A) ASAS 20; (B) ASAS 5/6; (C) ASAS 40. BIW, twice weekly; QW, once weekly. *p<0.05, etanercept 50 mg QW versus placebo. †p<0.05, etanercept 25 mg BIW versus placebo.
At week 12, partial remission occurred in a significantly greater proportion of patients receiving etanercept: 31.6% and 21.3% in the etanercept 50 mg-once-weekly and 25 mg-twice-weekly groups respectively, compared with 5.9% for the placebo group (p<0.05). In addition, onset of partial remission occurred significantly earlier for the etanercept groups than for the placebo group (p = 0.003 and p = 0.025, log rank test; for etanercept 50 mg-once-weekly and 25 mg-twice-weekly, respectively).
Improvement in physical function, as measured by the Bath Ankylosing Spondylitis Functional Index, was significantly greater for both the etanercept groups than for the placebo group at all time points (p<0.05), except at week 2 for the etanercept 25 mg-twice-weekly group.
Both etanercept treatment regimens significantly improved nocturnal back pain and physician global assessment of disease activity compared with placebo treatment from week 2 onwards (p<0.05); the improvements in the two etanercept groups were not significantly different from each other. As early as week 2, the CRP levels decreased significantly compared with baseline in the etanercept groups (p<0.001).
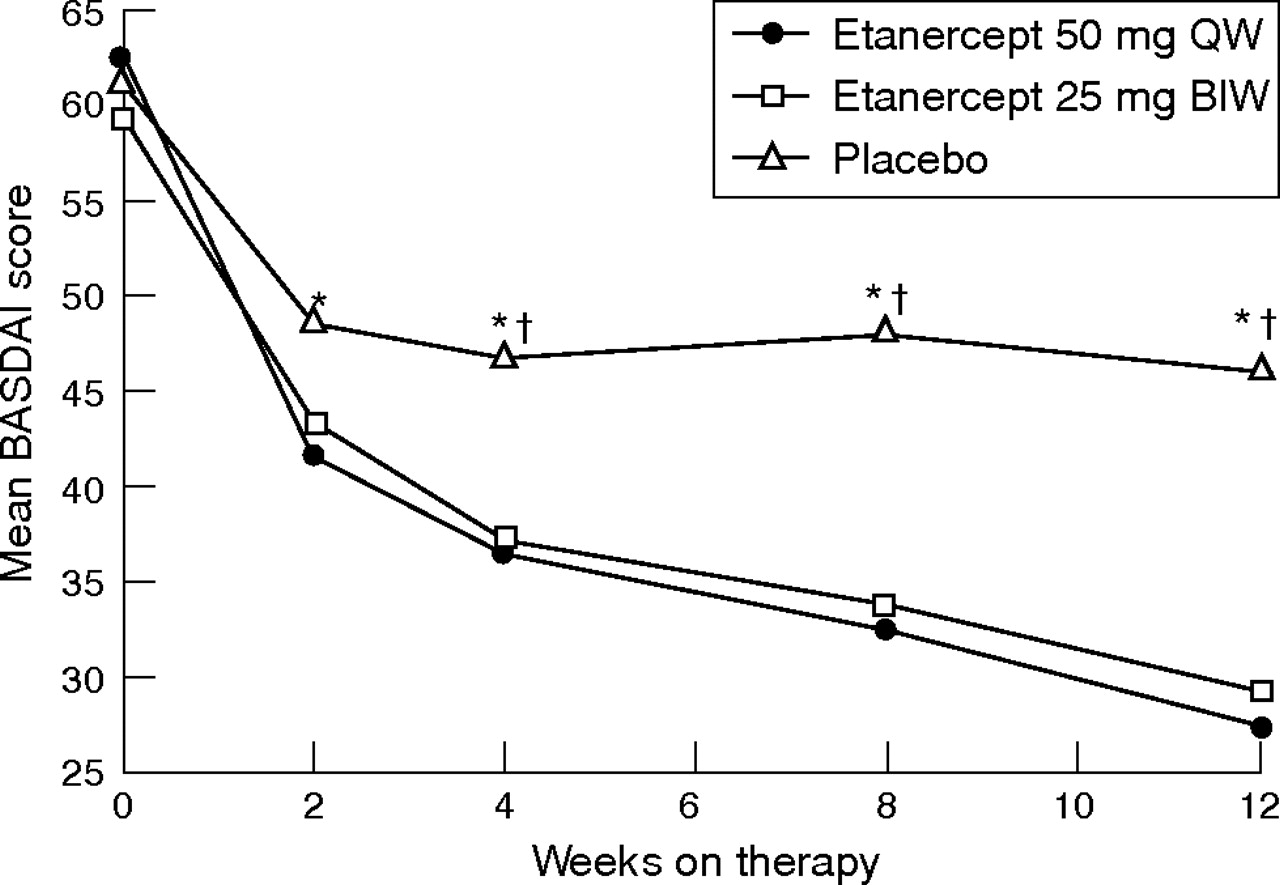
The mean changes from baseline in the BASDAI for both etanercept treatment groups over the 12 weeks showed significant improvement compared with the placebo group at most visits (fig 3). We found no significant difference between the two etanercept groups. The proportions of patients with at least 50% improvement in the BASDAI were significantly greater in the two etanercept groups compared with the placebo group at all time points (p<0.05) after week 2. At week 12, the proportions of patients achieving a 50% improvement in the BASDAI (60.0% and 58.0%, respectively, for etanercept 50 mg once weekly and 25 mg twice weekly, p = 0.729) were significantly greater than those receiving placebo (19.6%; p<0.001).
{kind=link}
{kind=link}
{kind=link}
Mean Bath Ankylosing Spondylitis Disease Activities Index (BASDAI) scores over time (modified intent-to-treat population; last-observation-carried-forward approach). BIW, twice weekly; QW, once weekly. *p<0.01, etanercept 50 mg QW versus placebo. †p<0.01, etanercept 25 mg BIW versus placebo.
The mean number of swollen and tender joints decreased in both etanercept-treated groups, but the difference reached significance only at some isolated time points. This could be due to the small numbers of active joints in the enrolled patients.
Spinal mobility was assessed using the modified Schober’s test, chest expansion and occiput-to-wall measurements. At 12 weeks, mean improvement from baseline in both etanercept groups was significantly higher than in the placebo group: 22.4% for etanercept 50 mg once weekly, and 25.4% for 25 mg twice weekly, versus 3.1% for placebo (p<0.001). Mean improvement in chest expansion for both etanercept groups at 12 weeks was greater than that for the placebo group, and was significantly greater in patients receiving etanercept 25 mg twice weekly (p = 0.024); improvement in occiput-to-wall measurement was 24.8% and 20.3% versus 7.7%, for etanercept 50 mg once weekly, 25 mg twice weekly and placebo, respectively (p<0.05 for etanercept 50 mg once weekly versus placebo).
Pharmacokinetics
We found no significant differences in the pharmacokinetic exposure between the two etanercept regimens. Mean (standard deviation (SD)) clearance was 0.068 (0.007) and 0.069 (0.006) l/h for patients with ankylosing spondylitis receiving etanercept 25 mg twice weekly (n = 148) and etanercept 50 mg once weekly (n = 154), respectively. Mean (SD) etanercept exposure at week 12, measured by the area under the curve (AUC), was 474 (121) and 466 (96) μg h/ml, respectively. The geometric least-squares mean AUC ratio of etanercept 25 mg twice weekly versus etanercept 50 mg once weekly was 99.4% (95.3–104%), and the 90% CI (95.3 to 104) fell within the bioequivalence range (80–125%), indicating that the etanercept 25 mg twice weekly and etanercept 50 mg-once-weekly dose regimens produced equivalent AUC exposures.
Safety
Patient exposure to etanercept was similar between treatment groups (32.6 and 31.5 patient-years for etanercept 50 mg once weekly and 25 mg twice weekly, respectively). Etanercept 50 mg once weekly or 25 mg twice weekly was generally well tolerated, with no unexpected safety findings. When comparing treatment-emergent adverse events reported at a frequency ⩾3% (table 3), injection site reactions were similar in the two etanercept groups and both were higher than those for placebo; back pain was higher in the placebo-treated group.
Patients with treatment-emergent adverse events (⩾3%)
The incidence of infections was similar among the three treatment groups; the most commonly occurring infections in all three groups were upper respiratory tract infections (table 3). We found no significant differences in reports of non-infectious serious adverse events among the three treatment groups (5.2%, 4.0% and 3.9% in the etanercept 50 mg-once-weekly, 25 mg-twice-weekly and placebo groups, respectively; p = 0.94). Two medically important infections, erysipelas (in the etanercept 50 mg-once-weekly group) and Streptococcus pyogenes (in the etanercept 25 mg-twice-weekly group) at an insulin catheter site, were reported in two patients (table 3).
No malignancies were reported, nor were any cases of multiple sclerosis, optic neuritis, aplastic anaemia, pancytopenia, demyelinating diseases or lupus. No patients had National Cancer Institute (NCI) grade 4 laboratory test result abnormalities; one patient (50 mg group) had NCI grade 3 total bilirubin and another patient (placebo group) had NCI grade 3 lymphocyte count, but neither discontinued because of these findings. At week 12, serum levels had normalised in the patient with raised bilirubin levels. The patient with the NCI grade 3 lymphocyte count had a pre-existing low lymphocyte count at screening with an NCI grade 3 value at weeks 4 and 12. Only two patients in each etanercept group had anti-etanercept antibodies, which were transient and non-neutralising.
At baseline, nine patients with a history of inflammatory bowel disease were enrolled. None of these patients had an inflammatory bowel disease-related adverse event during the study and no new cases were reported. No new diagnosis of uveitis was made in patients receiving etanercept; one case of uveitis was reported in the placebo group. Fifteen patients had prophylactic treatment for tuberculosis during the study, but none developed active disease.
No patients reported opportunist infections. One death (etanercept 25 mg-twice-weekly group) occurred after the study as a result of accidental injury.
DISCUSSION
The efficacy and safety of etanercept in the treatment of patients with ankylosing spondylitis have been evaluated in placebo-controlled studies.8,10–13 Our study, which has similar eligibility and disease severity criteria as in the pivotal trials of etanercept,8,10 is among the largest controlled trials with etanercept in patients with ankylosing spondylitis and the first to evaluate the efficacy and safety of etanercept 50 mg-once-weekly in the treatment of patients with this condition. This study was designed to assess the clinical equivalence and safety of etanercept 50 mg once weekly compared with the approved 25 mg-twice-weekly regimen in patients with ankylosing spondylitis. Both dose regimens were comparable in the treatment of patients with rheumatoid arthritis17; in Europe and the US, the etanercept 50 mg-once-weekly regimen has been approved as a treatment option for patients with rheumatoid arthritis.
The primary efficacy end point of the study was the proportion of ASAS 20 responders at week 12. This composite end point has been recommended by the international ASAS Working Group as a validated efficacy instrument in ankylosing spondylitis, and was the primary end point in two previous placebo-controlled clinical studies of etanercept.8,10 In the current study, the proportions of ASAS 20 responders at week 12 in the 50 mg-once-weekly group and the 25 mg-twice-weekly group were similar and considerably higher than those in the placebo group. Non-inferiority of etanercept 50 mg once weekly was achieved because the 95% CI for the proportion (%) difference between the two dose regimens, −7.1% to 12.9%, was well above the predefined non-inferiority margin of −18.5%. In addition, the proportion of ASAS 20 responders in this study compared favourably with that reported previously.8,10,11
In addition to the primary end point, we examined several other ASAS cut-offs, recommended for use in controlled trials of anti-TNF agents.20 As seen with the ASAS 20, markedly higher percentages of patients achieved at least 20% improvement in five of the six ASAS domains and at least a 40% improvement in three of four ASAS domains in the etanercept groups than achieved by the placebo group (p<0.001).
Examination of the components of ASAS response criteria verified the results obtained with the composite score. In addition, two independent measures of disease activity, BASDAI and CRP, also verified the improvement observed using the ASAS criteria. As observed in previous studies of etanercept in patients with rheumatoid arthritis, psoriatic arthritis and ankylosing spondylitis, the rapid decrease in CRP levels observed at the first post-dose visit at 2 weeks was maintained for the study duration. Limitation in spinal mobility, a major cause of disability in patients with ankylosing spondylitis, was also markedly improved in both etanercept treatment groups compared with the placebo group.
The efficacy results obtained with etanercept in this study on ankylosing spondylitis are consistent with those previously reported in a similar non-inferiority study on rheumatoid arthritis.17 In both conditions, a 50 mg per week dose of etanercept, given either as 50 mg once weekly or as 25 mg twice weekly, resulted in marked and sustained improvement in disease activity.
As previously seen in the non-inferiority study in patients with rheumatoid arthritis,17 the comparable efficacy profiles of the 50 mg-once-weekly and 25 mg-twice-weekly regimens were supported by the pharmacokinetic analysis. Both treatment regimens resulted in equivalent etanercept exposure in patients with ankylosing spondylitis. Thus, in these two rheumatic conditions, the pharmacokinetics of etanercept produced equivalent exposures for administration of either a once-weekly or a twice-weekly dose.
The two etanercept regimens were generally well tolerated in this study, with no unexpected safety findings in either group. The proportions of reported adverse events were similar between the two etanercept groups and not different from those reported in the placebo group. We found no significant differences in the incidences of infections or serious adverse events among the three groups.
In conclusion, the efficacy and safety of etanercept 50 mg once weekly was comparable with that of the standard regimen of 25 mg twice weekly in patients with ankylosing spondylitis. The pharmacokinetic analysis of the two dose regimens produced equivalent AUC exposures. No unexpected safety data were reported. Therefore, patients with ankylosing spondylitis can expect comparable clinical outcomes when treated with either etanercept 50 mg once weekly or 25 mg twice weekly.
Acknowledgments
We thank Ruth Pereira, PhD, Wyeth Clinical Publications Group, for assistance with preparation of the manuscript.
REFERENCES
Footnotes
-
Published Online First 28 September 2006
-
Etanercept Study 314 investigators: Claude-Laurent Benhamou, Orléans, France; Jürgen Braun, Herne, Germany; Marek Brzosko, Szczecin, Poland; Alain Cantagrel, Toulouse, France; Hanna Chwalinska-Sadowska, Warszawa, Poland; Laszlo Czirjak, Pécs, Hungary; Kurt De Vlam, Leuven, Belgium; Liana Euller-Ziegler, Nice, France; Flavio Fantini, Milano, Italy; Anna Filipowicz-Sosnowska, Warszawa, Poland; JS Hill Gaston, Cambridge, UK; Pal Geher, Budapest, Hungary; Piet Geusens, Diepenbeek, Belgium; Eugeniusz Kucharz, Katowice, Poland; Jolanta Lewandowicz, Lodz, Poland; Michel Malaise, Liège, Belgium; Emilio Martin Mola, Madrid, Spain; Neil McHugh, Bath, UK; Carlo Salvarani, Reggio Emilia, Italy; Jacques Sany, Montpellier, France; Lucas Settas, Thessaloniki, Greece; Jean Sibilia, Strasbourg, France; Fotini Skopouli, Athens, Greece; Zoltan Szekanecz, Debrecen, Hungary; Laszlo Tamasi, Miskolc, Hungary; Christiaan van Denderen, Amsterdam, The Netherlands; Jürgen Wollenhaupt, Hamburg, Germany; Henning Zeidler, Hannover, Germany; Irena Zimmermann-Gorska, Poznañ, Poland
-
Funding: This study was supported by Wyeth Pharmaceuticals, Collegeville, Pennsylvania, USA (study drug and grants to investigational sites).
-
Competing interests: DvdH was reimbursed by Wyeth, the manufacturer of etanercept, for attending several conferences and for running educational programmes, and has received research grants. JCdS was paid by Schering-Plough Pharma for running educational programmes and served as an advisory board member for Abbott. MD received grants from Wyeth to conduct clinical trials, and also fees for consulting and for speaking at symposia organised by Wyeth. PG was reimbursed by Wyeth, the manufacturer of etanercept, for attending a conference, and received fees for speaking at another conference. FR received fees for speaking to general practitioners who have been working with our hospital over the past 5 years. JS received fees for consulting and speaking, and funds for research. LP, JSW, SF and M-PB are employees of Wyeth Research. IvdH-B, XJ, IO, LS, JS and DW have no competing interests.