
Abstract
Vaccines against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) have shown high efficacy, but immunocompromised participants were excluded from controlled clinical trials. In this study, we compared immune responses to the BNT162b2 mRNA Coronavirus Disease 2019 vaccine in patients with solid tumors (n = 53) who were on active cytotoxic anti-cancer therapy to a control cohort of participants without cancer (n = 50). Neutralizing antibodies were detected in 67% of patients with cancer after the first immunization, followed by a threefold increase in median titers after the second dose. Similar patterns were observed for spike protein-specific serum antibodies and T cells, but the magnitude of each of these responses was diminished relative to the control cohort. In most patients with cancer, we detected spike receptor-binding domain and other S1-specific memory B cell subsets as potential predictors of anamnestic responses to additional immunizations. We therefore initiated a phase 1 trial for 20 cancer cohort participants of a third vaccine dose of BNT162b2 (NCT04936997); primary outcomes were immune responses, with a secondary outcome of safety. At 1 week after a third immunization, 16 participants demonstrated a median threefold increase in neutralizing antibody responses, but no improvement was observed in T cell responses. Adverse events were mild. These results suggest that a third dose of BNT162b2 is safe, improves humoral immunity against SARS-CoV-2 and could be immunologically beneficial for patients with cancer on active chemotherapy.
Similar content being viewed by others


Main
The Coronavirus Disease 2019 (COVID-19) pandemic has led to over 200 million infections worldwide and claimed over 4 million lives to date. Although non-pharmaceutical public health interventions have managed to control outbreaks in some countries, most of the global population will depend upon vaccines to mitigate the pandemic. Since the identification in January 2020 of SARS-CoV-2 as the causative agent of COVID-19 (refs. 1,2), vaccines with very high efficacy have been developed and deployed with remarkable speed. Independent clinical trials demonstrated 94–95% vaccine efficacy against symptomatic disease caused by SARS-CoV-2 for both the Pfizer/BioNTech and Moderna mRNA-based vaccines3,4. On the basis of these data, in December 2020, both the Pfizer/BioNTech and Moderna vaccines were granted emergency use authorization by regulatory agencies in North America.
These clinical trials, however, largely excluded immunocompromised individuals, including patients on immunosuppressive therapies to control chronic inflammatory conditions, patients with primary immunodeficiencies, recipients of organ transplants and patients with cancer on cytotoxic chemotherapy. Concern has been particularly high about the effect of COVID-19 on patients with cancer because a study of 928 patients from the COVID-19 Cancer Consortium showed a 13% 30-d all-cause mortality from COVID-19, which is 10–30 times greater than that observed in the general population5. Importantly, the investigators noted a higher risk of death in patients with active cancer5.
Several recent reports have shown diminished immune responses to SARS-CoV-2 infections and mRNA vaccines in subsets of immunocompromised patients, although these vary greatly with the nature of the immunosuppressive therapy6,7,8,9. For example, patients with autoimmune conditions or chronic lymphocytic leukemia treated with B cell-depleting antibodies have predictably diminished humoral responses to vaccination, whereas responses by patients on anti-TNF therapies are less affected6,7. Notably, organ transplant recipients mount very poor antibody responses to the first mRNA immunization relative to healthy individuals10, which increase somewhat after the second immunization8. Similarly, in patients with cancer with solid or hematological malignancies, antibody responses are markedly diminished after the first immunization but improve somewhat after the second9. More data are required to determine whether additional immunizations might further protect this vulnerable population.
Here we report on the serological and cellular immune responses after two-dose BNT162b2 vaccination of patients with solid tumors on active cytotoxic chemotherapy compared to healthy controls. We also report on the outcomes of a phase 1 trial of a third vaccine dose subsequently initiated in the cancer cohort based on our initial two-dose results.
Results
Participant characteristics of control and cancer cohorts
For the observational study (Table 1), 53 patients with a known diagnosis of a solid tumor malignancy who were on active immunosuppressive cancer therapy were enrolled through the University of Arizona Cancer Center during their routine care. The 50 participants in the control cohort of the observational study were enrolled through the State of Arizona’s COVID-19 BNT162b2 vaccine point of distribution site at the University of Arizona during the phase 1B vaccination program while in the observational waiting area after their first dose. Table 1 summarizes chemotherapeutic regimens prescribed for the patients in the cancer cohort, with a full list in Supplementary Table 1. All eligible patients in the cancer cohort were subsequently invited to participate in an interventional trial to receive a third dose of vaccine (NCT04936997). Twenty patients were consented and participated in this interventional trial (Extended Data Fig. 1). The primary endpoint for both of these studies was change in neutralizing antibody titers; all other immune biomarkers were secondary endpoints. Exploratory analyses include hierarchical clustering and age-adjusted analyses.
Antibody responses to BNT162b2 vaccination
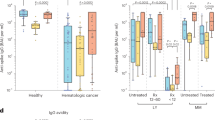
Blood samples for serological and cellular analyses were collected several minutes after the first immunization, at the time of the second immunization 3 weeks later and again 5–11 d after the second vaccination (Fig. 1a). Peripheral blood mononuclear cell (PBMC) counts were similar between the cancer and control cohorts (Extended Data Fig. 2a and Supplementary Fig.). However, we noted a significant reduction in the frequency of CD19+ B cells and an increase in CD13+ myeloid cells in the cancer cohort relative to controls (Extended Data Fig. 2b,c). Despite the overall reduction in the frequency of CD19+ B cells in the cancer cohort, naive and other activated subsets were well-represented within these B cells (Extended Data Fig. 2d and Supplementary Fig.). Using serum from each of these samples, we first obtained single-dilution semi-quantitative data on spike protein-specific antibody levels. Of control cohort participants, using a University of Arizona clinical serology test11, four tested positive for prior SARS-CoV-2 exposure at the first blood draw and were excluded from further analyses. Four cancer cohort participants self-reported prior COVID-19 but did not display serological evidence of prior infection and were not excluded. For both the control and cancer cohorts, we observed progressive increases after the first and second vaccinations in antibodies specific for the S2 region of the spike protein (Fig. 1b). This region contains several antibody epitopes that are conserved across other common human β-coronaviruses12,13,14,15,16, including at least one weakly neutralizing epitope17,18. Although both the cancer and control cohorts showed responses, median S2-specific antibody values were diminished in patients with cancer relative to the control cohort at matched time points (Fig. 1b). As most neutralizing and protective antibodies are directed to the receptor-binding domain (RBD) of the spike protein19,20, we also semi-quantitatively determined the relative levels of these antibodies. Increases were also seen for RBD antibodies in both the healthy and cancer cohorts after each vaccination (Fig. 1c). However, as with antibodies against S2, the levels of RBD antibodies at draws 2 and 3 after vaccination in the cancer cohort were diminished relative to healthy controls (Fig. 1c). To obtain more quantitative information, we performed a full dilution series to determine antibody titers against RBD (Extended Data Fig. 2e). Consistent with the semi-quantitative results, RBD antibody titers increased after the second immunization in both groups, but the median titers observed in the cancer cohort were reduced by more than 11-fold relative to healthy controls (Fig. 1d). Seven of the cancer cohort, but none of the control cohort, failed to generate RBD-specific antibody titers above the limit of detection.

a, Schematic of blood collection (draws) after vaccination. b, Semi-quantitative 1:40 serum dilution ELISA results for reactivity to the S2 region of the SARS-CoV-2 spike protein. Lines connect the same individual across time points. Repeated-measures ANOVA examines the differences in slopes between cohorts, independently from the mean differences that were demonstrated at draw 3 between cohorts. There is a statistically significant difference in slopes between cancer and control cohorts (P < 0.0001), and the average rate of change is increasing at a steeper rate in the control cohort. These paired rates between draws by cohort are statistically different in the control cohort compared to the cancer cohort for both draws 1 and 2, although it is not different between draws 2 and 3 (P < 0.0001 and P < 0.2945, respectively). c, Semi-quantitative 1:40 serum dilution ELISA results for reactivity to the RBD of the SARS-CoV-2 spike protein. Lines connect the same individual at each draw. There is a statistically significant difference in slopes between cancer and control cohorts (P < 0.0001), and the average rate of change is steeper in the control cohort. These paired rates between draws are statistically different in the control cohort compared to the cancer cohort for both draws 1–2 and draws 2–3 (P < 0.0001 and P < 0.0043, respectively). d, Quantitative titers of RBD antibodies in control and cancer cohorts. A serum concentration beginning at 1:80 was serially diluted 1:4, and AUC values were calculated. Lines connect the same individual across time points. There is a statistically significant difference between draws 2 and 3 between cancer and control cohorts (P < 0.0001), and the average rate of change is at a steeper increase in the control cohort. For b–d, two-sided P values from t-test statistics were calculated for pairwise differences using two-way ANOVA. Post hoc testing for multiple comparisons between draws was performed using Sidak’s correction. Comparisons were made either within cohorts or between cohorts at each draw; P values greater than 0.05 are not depicted. All data points represent biological replicates (n = 50 for the control cohort and n = 53 for the cancer cohort). Abs, antibodies.
For most vaccines, neutralizing antibody titers are the best correlate of protection from infections. We, therefore, directly assessed antibody-mediated neutralization of authentic live SARS-CoV-2 (WA1 isolate) after the first and second immunizations. After the first shot, we observed a median 90% plaque reduction neutralization test (PRNT90) titer of 60 in the control cohort and 20 in the cancer cohort (Fig. 2). However, whereas all but one participant in the control cohort showed detectable virus neutralizing activity, this was observed in only 67% of the cancer cohort (Fig. 2). After the second immunization, all healthy controls had virus-neutralizing antibodies, with a median PRNT90 titer of 540 (Fig. 2). By contrast, 80% of the cancer cohort had detectable neutralizing antibodies, with a median titer of 60 (Fig. 2). Virus-neutralizing titers correlated with overall RBD-specific antibodies (Extended Data Fig. 3). These results demonstrate that most of the cancer cohort generated protective antibodies but at levels well below that of the control cohort after the second vaccine dose. We did not find any obvious clinical characteristics that would have modified the relationship between immunosuppression and vaccine response. Of the non-responders, 60% were patients with breast cancer, 90% were female and the median age was 64 years. Although there is no statistical power to compare this subgroup to the overall cancer cohort, the only obvious difference was treatment timing (Table 1), as the average time between treatment and vaccine dose 2 was over 2 weeks in the overall group compared to less than 1 week in non-responders.

Virus neutralization assays were performed using the WA1 isolate of SARS-CoV-2. Serial 1:3 dilutions of serum were performed and tested for the ability to prevent plaques on Vero cells. The lowest concentration capable of preventing more than 90% of plaques was considered to be the PRNT90 value. Example images are shown for the control and cancer cohorts, with the red box indicating the PRNT90 titer. Quantification is shown below. Lines connect the same individual across time points. There is a statistically significant difference between draw 2 and draw 3 between cancer and control cohorts (P < 0.0001), and the average rate of change is increasing at a steeper rate in the control cohort (P = 0.0002). Two-sided P values from t-test statistics were calculated for pairwise differences using two-way ANOVA. Post hoc testing for multiple comparisons between draws was performed using Sidak’s correction. Comparisons were made either within cohorts or between cohorts at each draw; P values greater than 0.05 are not depicted. All data points represent biological replicates (n = 50 for the control cohort and n = 53 for the cancer cohort).
T cell responses to BNT162b2 vaccination
Previous studies found that potentially protective T cell responses can be observed in convalescent individuals with COVID-19 and in animal models when antibody levels are very low, such as after asymptomatic infections21,22,23,24. To quantify T cell responses in our healthy and cancer cohorts, PBMCs were cultured overnight with either activating anti-CD3 antibodies (Extended Data Fig. 4a) or a pool of overlapping spike protein peptides capable of presentation on both HLA-I and HLA-II (Fig. 3a). Enzyme-linked immune absorbent spot (ELISpot) assays were then performed to quantify interferon gamma (IFNγ)-producing T cells relative to paired control wells in which no peptides were added. In the control cohort, we observed a marked increase in the median frequency of IFNγ+ T cells after the first vaccination relative to pre-vaccination time points (2.9-fold, P = 0.02) and a further increase after the second vaccination (2.6-fold, P = 0.0003) (Fig. 3a). In the cancer cohort, the first vaccination did not induce a statistically significant increase in the median frequency of spike-specific IFNγ+ T cells at draw 2, but a clear increase was observed at draw 3 (4-fold, P = 0.0007) (Fig. 3a), although there was substantial variability in the response. T cell frequencies in the cancer cohort approached, but remained below, the levels observed in the control cohort after the second vaccination (P = 0.03; Fig. 3a). To estimate CD8+ and CD4+ T cell contributions, we re-tested samples with the highest spike-specific T cell frequencies in the presence or absence of blocking antibodies against HLA-I and/or HLA-II, respectively. Substantial variation was observed across individuals in both the control and cancer cohorts, especially in HLA-I-dependent CD8+ T cell responses (Extended Data Fig. 4b). Nonetheless, the data indicate that most individuals mount both CD4+ and CD8+ T cell responses.

a, PBMCs were cultured for 24 h in the presence or absence of a pool of overlapping spike protein peptides. IFNγ-producing cells were quantified by ELISpot. Example images are shown for the control and cancer cohorts at time points 1 and 3. Quantification is shown below of the no-peptide background-subtracted data. Lines connect the same individual across time points. There is a statistically significant difference in slopes between cancer and control cohorts (P = 0.0284), and the average rate of change is increasing steeper in the control cohort. Although the overall rate of change was significant (draw 1 to draw 3, P = 0.0455), the rates of change between draw 1 to draw 2 and draw 2 to draw 3 were not statistically significant (P = 0.0642 and P = 0.9891, respectively). The inability to detect a statistical difference, particularly between draw 1 and draw 2, is likely due to sample size and variability, as the cancer cohort difference is flatter than the cancer cohort between these two draws; these analyses were performed on a log-transformed scale. Two-sided P values from t-test statistics were calculated for pairwise differences using two-way ANOVA. Post hoc testing for multiple comparisons between draws was performed using Sidak’s correction. Comparisons were made either within cohorts or between cohorts at each draw; P values greater than 0.05 are not depicted. All data points represent biological replicates (n = 50 for the control cohort and n = 53 for the cancer cohort). b, Draw 1 spike-specific T cell frequencies were subtracted from draw 3 frequencies as calculated in a and plotted as a function of PRNT90 titers. Frequencies of individuals with detectable spike-specific T cells are shown above each group. All data points represent biological replicates (n = 53).
To determine whether participants with poor neutralizing antibody titers might be partially protected by T cell responses, we examined T cell frequencies grouped by neutralizing antibody titers. Spike protein peptide-specific T cell frequencies at draw 1 were subtracted from the final draw 3 numbers to define individuals who mounted a response to vaccination. As has previously been described in post-infection responses21,25, spike protein peptide-specific T cell frequencies correlated relatively poorly with neutralizing antibody titers for both the healthy and cancer cohorts (Fig. 3b). These data revealed that 4/10 patients with cancer had detectable T cell responses even when PRNT90 titers were undetectable (Fig. 3b). Thus, despite chemotherapy-induced immune suppression, relatively few patients with cancer failed to make any detectable neutralizing antibody or T cell response. Nonetheless, these responses were substantially diminished relative to the control cohort, likely due to anti-cancer therapy.
One drawback to these interpretations is that the median age of the cancer cohort was greater than that of controls (Table 1). This raises concerns that some of the differences that we observed were effects of age rather than of anti-cancer therapy. The only immunological parameter that showed an age-dependent effect was anti-RBD antibody levels, which did show a decline with increasing age in the control cohort (Pinteraction = 0.01; Extended Data Fig. 5a). However, we observed no such age-dependent differences in the cancer cohort. Moreover, no other immunological parameters, such as neutralizing antibody levels or T cell responses, were altered as a function of age (Extended Data Fig. 5b,c). Furthermore, when limiting the data to participants older than 39 years (upper three quartiles), the differences between the two cohorts remained statistically significant for all immunological parameters (RBD antibodies, P < 0.0001; neutralizing antibodies, P < 0.0001; spike-specific T cells, P = 0.04). These data are consistent with the only modest age-dependent effects in immune responses reported in BNT162b2 phase 1 clinical trials26.
Because we assessed responses between 5 and 11 d after the second immunization, we examined whether there were any time-dependent changes in responses within this window. RBD-specific antibodies, neutralizing titers and T cell responses did not obviously differ as a function of time after vaccination (Extended Data Fig. 6a). Most control and cancer cohort participants were tested at 7–8 d after vaccination (Extended Data Fig. 6a). Although the study was not powered for subgroup analyses, we also examined whether the tumor subtype might influence immune responses. No obvious differences were observed in antibody and T cell responses among breast, pancreatic and other tumor types (Extended Data Fig. 6b). We noted that one participant in the cancer cohort mounted a much stronger antibody response than the rest of the group (Extended Data Fig. 6a,b). This participant had self-reported prior COVID-19 infection, despite seronegativity before the first immunization. However, three other cancer cohort participants with self-reported prior COVID-19 showed no unusual patterns of antibody or T cell responses (Extended Data Fig. 6c). Based on our initial inclusion criteria of seronegativity before vaccination, we retained these individuals in our analyses, but we note that at least one of these four participants might be mounting recall, rather than primary, responses. Together, these data suggest that anti-cancer therapy hampers immune responses to the COVID-19 vaccine BNT162b2. The effectiveness of these diminished immune responses in preventing COVID-19 is presently unknown.
Memory B cell subsets after BNT162b2 vaccination
Memory B cell frequencies are predictive of anamnestic responses after booster vaccination27 and, presumably, viral exposures. We first quantified RBD-specific CD19+ B cells in the control and cancer cohorts using antigen tetramers (Fig. 4a). We also simultaneously measured memory B cells that bound the S1 region of the spike protein but not the RBD (Fig. 4a), as the N-terminal domain of S1 contains a potent neutralizing epitope28,29. S2 tetramer reagents yielded high flow cytometric background staining of naive B cells and myeloid cells and were thus not used to further define antigen-specific memory lymphocytes (data not shown). Within the control cohort, a clear increase in total RBD-specific B cells was observed after vaccination, but no statistically significant increase was observed in such cells in the cancer cohort (Extended Data Fig. 7). Neither the control cohort nor the cancer cohort showed a significant increase in S1-specific B cells with vaccination (Extended Data Fig. 7).

a, Example gating strategy of RBD- and S1-specific CD21− isotype-switched memory B cells (full gating strategy is shown in Supplementary Fig.). b, Quantification of memory B cell and plasmablast subsets after vaccination. Isotype-switched memory B cells expressing or lacking CD21 are shown in plots along with plasmablasts. Cells that bind both RBD and S1 are annotated as RBD+, whereas cells that are specific only for S1 are denoted as S1+. Lines connect the same individual across blood draws; analyses were done on the arcsin of the square root transformation, to standardize the small percentages. There is a statistically significant difference in slopes between cancer and control cohorts for CD21− RBD+ and S1+ (P < 0.0001 and P < 0.0001, respectively), and the average rate of change is increasing in the control cohort for both CD21− RBD+ and S1+, but the slope increases more dramatically between draws 2 and 3 in the control cohort compared to the cancer cohort, which has a modest linearly increasing slope. The average rate of change was statistically significantly different for CD21+ RBD+ (P < 0.0001), with a linearly increasing slope in the control cohort that is much steeper than in the cancer cohort. There was no statistically significant difference in slopes between cancer and control cohorts for CD21+ S1+ (P = 0.1959). Two-sided P values from t-test statistics were calculated for pairwise differences using two-way ANOVA. Post hoc testing for multiple comparisons between draws was performed using Sidak’s correction. Comparisons were made either within cohorts or between cohorts by draw; P values greater than 0.05 are not depicted. All data points represent biological replicates (n = 50 for the control cohort and n = 53 for the cancer cohort). c, RBD-specific DN2 and DN3 and S1- and RBD-specific isotype-switched CD21− memory B cells were added for the cancer cohort. Summed draw 1 memory B cell frequencies were subtracted from the summation of draw 3 frequencies for each individual. These values were grouped by PRNT90 titers. Frequencies of individuals with detectable memory B cells are shown above each group. All data points represent biological replicates (n = 53). Sw, switched.
To gain more resolution, we examined antigen-specific frequencies within defined memory B cell subsets. These subsets exhibit different behaviors in recall responses, generating either plasmablasts or new germinal centers30,31,32,33,34,35,36. These lineage potentials correlate with antibody isotype and other markers30,31,32,33,34,35,36. We, therefore, quantified RBD- and S1-specific naive B cells, plasmablasts and memory B cell subsets after vaccination35,37,38,39. These subsets include IgG+ and IgM+ CD27+CD21+ classical resting memory B cells, CD27+CD21−CD11c+ pre-plasmablast memory B cells, CD27−IgD−CD11c−CD21+ DN1 cells, CD27−IgD− CD11c+ DN2 cells and CD27−IgD−CD11c−CD21− DN3 cells (Fig. 4a and Supplementary Fig.). In the control cohort, we observed a clear increase in the frequency of isotype-switched RBD-binding CD21+ classical resting memory B cells as well as CD21− pre-plasmablast memory B cells after each vaccination (Fig. 4b). Isotype-switched S1-binding CD21− memory B cells were also observed to increase after each vaccination of the control cohort (Fig. 4b). After the second vaccine dose in the cancer cohort, we also observed increases in the mean frequencies and/or rates of change of isotype-switched RBD- and other S1-specific pre-plasmablast CD21− memory B cells, but the median levels were ~tenfold lower than those observed in the control cohort (Fig. 4b). We were unable to detect isotype-switched RBD- or S1-specific classical resting memory B cells above pre-vaccination levels in the cancer cohort (Fig. 4b). Some other RBD- and S1-binding memory B cell subsets were detectable in the healthy and cancer cohorts, but, in general, the frequencies of these cells were substantially lower than the isotype-switched CD27+ subsets and not consistently increased with each immunization (Extended Data Fig. 8). We were unable to detect antigen-specific cells above background levels in naive B cells (Extended Data Fig. 8). Thus, RBD- and S1-specific B cells early after vaccination are enriched in IgG+ memory subsets. These cells are biased toward plasma cell fates30,35, although secondary germinal centers could conceivably arise from classical CD21+ memory cells34. In both cohorts, we observed increases in RBD-specific antibody-secreting plasmablasts, with no statistically significant differences between cohorts (Fig. 4b). S1-specific plasmablasts were not readily apparent in either group (Fig. 4b). This might be partly due to poor survival of plasmablasts after freezing and partly due to lower surface B cell receptor levels40.
We next examined whether RBD- and S1-specific memory B cells could be detected in patients with cancer with no or low levels of neutralizing antibodies. Previous studies showed that memory B cell numbers and specificities correlate only modestly with serum antibodies36,41,42,43,44. CD21− RBD+ and S1-specific memory B cell frequencies at draw 3 were added to DN2 and DN3 RBD+ memory B cell frequencies for each patient with cancer, as these subsets were the only ones in which patients with cancer consistently showed vaccine-induced increases (Fig. 4b and Extended Data Fig. 8). Next, the corresponding pre-vaccination draw 1 frequencies were subtracted to correct for background levels in each patient. These net memory B cell frequencies were then plotted as a function of virus neutralization titers. Patients without detectable neutralizing antibodies also generally lacked RBD- and S1-specific memory B cells (Fig. 4c). By contrast, patients with modest but detectable neutralizing antibody titers consistently showed RBD- and S1-specific memory B cells after the second immunization (Fig. 4c). These data suggest that patients with low but detectable spike-specific antibodies would likely generate anamnestic responses after a third immunization, conceivably approaching levels seen in healthy controls after the second vaccination.
Responses of patients with cancer to a third dose of BNT162b2
To directly determine whether and how immunity can be improved by a third vaccination, we initiated an interventional trial for our cancer cohort. Twenty of the original cohort agreed to participate and met the inclusion criteria (Methods). Participants were not informed of their immune response results. There were no statistically significant associations between participation and draw 3 RBD-specific antibodies, neutralization titers or spike-specific T cells. However, we note that the study was not powered sufficiently to specifically preclude these differences. All patients were contacted within 2–4-week windows to inquire about adverse events (AEs). No serious adverse events (SAEs) were noted (Fig. 5a), with nine (45%) participants experiencing injection site pain. Other minor AEs included generalized myalgia (15%), bone pain (5%), fatigue (10%), chills (10%) and appetite loss (5%). There were no obvious demographic differences between the 20 participants and the original cancer cohort; however, these participants did have a shorter window between administration of cancer treatment and blood draws for analyses. Patients in this cohort had gastrointestinal cancers predominantly (75%), compared to 51% in the original cancer cohort; the remaining five participants (25%) had a breast cancer diagnosis, compared to 42% in the original cancer cohort.

a, Frequencies of AEs after third vaccine dose. b, RBD-specific antibody titers were quantified at the time of the third immunization (draw 4) and 1 week afterwards (draw 5). Data from draw 3 are the same as those in Fig. 1d and are shown again for context. The blue box plot shows median and boundaries between the 1st and 3rd quartiles for the control cohort at draw 3; whiskers depict 5th–95th percentiles. All data points represent biological replicates (n = 20). P values are based on two-sided paired t-tests from the interventional cancer cohort (n = 20) based on draw 4 versus draw 5 comparisons; draw 3 cancer cohort (n = 53) and control cohort (n = 50) data are for descriptive comparison only. c, Neutralizing antibody titers were quantified at the time of the third immunization (draw 4) and 1 week afterwards (draw 5). Data from draw 3 are the same as those in Fig. 2. The blue box plot shows median and boundaries between the 1st and 3rd quartiles for the control cohort at draw 3; whiskers depict 5th–95th percentiles. All data points represent biological replicates (n = 20). P values are based on two-sided paired t-tests from the interventional cancer cohort (n = 20) based on draw 4 versus draw 5 comparisons; draw 3 cancer cohort (n = 53) and control cohort (n = 50) data are for descriptive comparison only. d, Spike-specific T cell responses were quantified at the time of the third immunization (draw 4) and 1 week afterwards (draw 5). Data from draw 3 are the same as those in Fig. 3a. The blue box plot shows median and boundaries between the 1st and 3rd quartiles for the control cohort at draw 3; whiskers depict 5th–95th percentiles. All data points represent biological replicates (n = 20). P values are based on two-sided paired t-tests from the interventional cancer cohort (n = 20) based on draw 4 versus draw 5 comparisons; draw 3 cancer cohort (n = 53) and control cohort (n = 50) data are for descriptive comparison only. Abs, antibodies.
Blood samples were collected at the time of the third vaccine dose (draw 4) and 1 week afterwards (draw 5) (Extended Data Fig. 9). RBD-specific antibodies, virus-neutralizing antibodies and spike-specific T cells were quantified. A modest but consistent and statistically significant increase from draw 4 to draw 5 was observed in mean RBD-specific antibody titers (0.49 versus 0.72, P = 0.0005; Fig. 5b). This was accompanied by a threefold increase in median virus-neutralizing antibody titers (60 versus 180, P = 0.01; Fig. 5c). Interestingly, two participants who had no detectable neutralizing antibodies at draw 3 showed an increase at draw 4, even before the third immunization (Fig. 5c). In both cases, neutralizing antibodies increased further after the third shot (Fig. 5c). We observed no overall increase in T cells after booster immunization of the cancer cohort (Fig. 5d). Because participants received the third shot between 42 and 111 d after the third blood draw, we examined whether the duration of time between immunizations might influence the magnitude of the antibody response, as has been reported for doses 1 and 2 (ref. 45). However, we observed no correlation between the time between doses and the magnitude of the RBD-specific or neutralizing antibody recall responses in this small cohort (Extended Data Fig. 9b,c).
Previous studies of vaccinations of convalescent individuals with COVID-19 revealed a strong correlation between pre-existing memory B cells and the magnitude of the antibody response after immunization27. To determine whether such a relationship could be observed in our cancer cohort, we plotted the draw 3 RBD-specific memory B cell frequencies (switched CD21−, switched CD21+, DN2 and DN3) against the change in RBD-specific antibodies after the booster immunization. Unexpectedly, we observed no correlation between these parameters (Extended Data Fig. 9d). A similar lack of correlation was observed between summed RBD and S1-specific memory B cells and boosted virus-neutralizing antibody titers (Extended Data Fig. 9e). These data suggest that, unlike in healthy individuals27, memory B cell frequencies are not quantitatively predictive of the magnitude of the antibody response in our cancer cohort.
To begin to explain this lack of correlation between memory B cells and subsequent anamnestic responses, we performed comparisons of memory B cell frequencies, antibody levels and T cell responses before the third vaccine dose at draw 3 (Extended Data Fig. 10). Most parameters were well-correlated with each other in the control cohort in biologically rational ways. For example, isotype-switched CD27+ and DN2 subsets clustered together and were highly correlated (Extended Data Fig. 10). Within the cancer cohort, however, memory B cell subsets were not well-correlated and did not cluster with each other (Extended Data Fig. 10). This suggests a lack of coordination between aspects of the response that are normally linked, which, in turn, might lead to quantitatively unpredictable recall responses. Nonetheless, antibody responses did improve in most patients with cancer who received a third vaccine dose.
Discussion
The COVID-19 pandemic has dramatically affected the world, with a profound effect on patients with cancer and their care. Thus, the development of COVID-19 vaccines was anxiously awaited in the cancer community. Given that none of the COVID-19 vaccine trials included patients with active malignancies3, the efficacy of these vaccines in patients with solid tumors who are on active therapy was not reported. Although previous studies in patients with colorectal and breast cancers on active chemotherapy who received influenza vaccination show that patients can mount a serological response, the immunogenicity of the COVID-19 vaccines in these patients is largely unknown46,47. A recent study of 658 organ transplant recipients showed a lower antibody response after two doses of the BNT162b2 COVID-19 vaccine when compared to immunocompetent study participants8. Similarly, one study suggested that patients with cancer do not mount the same antibody responses to the BNT162b2 vaccine as healthy controls9.
Our results agree with certain aspects of these findings: as with a recent study of immunocompromised patients with cancer9, we observed lower overall antibody and T cell responses in patients with cancer compared to control cohorts. However, in contrast to the previous results, we observed that most of our patients seroconverted after the first immunization, as measured by live virus neutralization assays. This frequency further increased after the second vaccination. These differences could potentially be explained by the nature of our cohort, which did not include patients on immunotherapy or patients with hematologic malignancies. In addition, neutralization assays using authentic viral isolates, as we used here, tend to be more sensitive than experiments performed with spike protein-pseudotyped lentiviruses11,48.
It is encouraging that we also observed T cell responses in most vaccinated patients with cancer, including nearly half who mounted undetectable neutralizing antibody responses. Unlike antibodies, these T cell responses were only modestly reduced relative to the control cohort. Of note, many individuals possess spike-reactive memory T cells, but not B cells or antibodies, even before SARS-CoV-2 exposure or vaccination49,50,51,52,53. It is possible that these pre-existing coronavirus cross-reactive memory T cells dominate after vaccination, diminishing the negative effect of anti-cancer therapy on immunization. The resulting CD4+ T cells could potentially help naive B cells participate in subsequent responses to vaccines and infections, which might help explain the poor correlations that we observed between memory B cell frequencies and the magnitude of the recall responses in patients with cancer. Given that T cells reduce viral loads and disease even when neutralizing antibody levels are low21,23,24,54, these data suggest that vaccination might confer at least partial protection and reduce the likelihood of severe COVID-19 in most patients with cancer.
Nonetheless, when compared to individuals not on immunosuppressive therapy, the magnitudes of vaccine-induced antibody and T cell responses were substantially reduced in patients with cancer. These reduced levels might be particularly problematic when faced with variants possessing some neutralizing antibody-evading mutations, such as beta, gamma or delta55,56. Some participants in our cohort failed to mount detectable antibody or T cell responses by 1 week after immunization, although several did show improvement over time. This seems likely to diminish vaccine effectiveness relative to the benchmark of 94–95% in non-immunocompromised populations3,4.
Several recent studies reported improved antibody responses in transplant recipients after a third dose, although neutralizing antibodies and T cells were not quantified57,58. We therefore initiated a trial to determine whether a third immunization would improve immunity in our cohort on active anti-cancer therapy. Interestingly, two participants who initially failed to mount detectable responses by 1 week after the second vaccination later displayed detectable antibodies before the third dose—one patient with cholangiocarcinoma and another with pancreatic cancer. This suggests that, for at least a subset of the non-responding cancer cohort, antibody responses might be delayed but not completely absent. After the third immunization, neutralizing antibody levels improved in 16 of 20 participants. In all but one of the participants who improved, neutralizing antibody titers reached 180 or greater. In non-human primate and modeling studies, this level is protective against disease24,59,60. Nonetheless, the overall antibody increases induced by the booster immunization were fairly modest, and, for reasons that are unclear, no further improvement was observed of circulating spike-specific T cell frequencies.
Our cancer cohort naturally had an expected heterogeneity in terms of cancer diagnoses, the types of cytotoxic therapy that patients received and the timing of these therapies relative to vaccine dose. Thus, it is difficult to draw conclusions related to which solid tumors were associated with a better vaccine response or which therapies correlated with the non-responders. However, it is worth noting that most of the initial non-responders had blood collected for immune analysis 7–14 d after their most recent treatment with cytotoxic agents. This time course is aligned with a nadir in blood counts and the peak of myelosuppression from traditional chemotherapy agents. Although the numbers are too small to draw strong conclusions, these findings are certainly hypothesis-generating and merit further exploration to better understand the ideal timing for vaccination in patients on active immunosuppressive therapy. Our cancer cohort was also, on average, older than participants in the control cohort. There did appear to be an age-moderated effect within the control group on anti-RBD titers, which, in turn, could affect the magnitude of the differences that we observed between the control and cancer cohorts. However, no other immunological parameters were similarly affected, and we did not observe age-moderated effects within the cancer cohort for any immunological parameter. Thus, the major driver of diminished responses in the cancer cohort is likely to be anti-cancer therapy rather than age.
Together, our data suggest that most patients with cancer on active chemotherapy are likely to exhibit improved antibody levels, which has been correlated with protection against disease59,60, after a third immunization. However, given the relatively modest increases in antibodies and recalcitrance of T cells, expectations should remain tempered as to the degree of benefit. Quantitative antibody tests can potentially be used to select individuals who need, and would most benefit from, a booster.
Methods
Participant selection
Recruitment for both the healthy cohort and the cancer cohort was approved by the University of Arizona Institutional Review Board (IRB) in January 2021. The interventional amendment to the protocol and subsequent enrollment for a third shot in patients with cancer was activated in June 2021 (NCT04936997). Participants were recruited to the control cohort during the phase 1B Pima County COVID-19 vaccine rollout. Participants scheduled for vaccine appointments at the Banner University Medical Center North site were approached with the IRB-approved consent and sequentially enrolled. Thereafter, patients with a cancer diagnosis were enrolled at the University of Arizona Cancer Center. Informed consent was obtained for all participants, and participants were aware that they would not be informed of their specific immune results. No compensation was offered for participation. Eligible patients with solid tumors had to have active disease and be receiving ongoing cytotoxic systemic therapy. Patients receiving immunotherapy were excluded. Demographic information was collected, in addition to cancer diagnosis and type of anti-cancer therapy. Dates of last treatment before vaccine administration were also noted. In total, 73 control cohort participants were consented, and 65 completed all three blood draws and both vaccine shots; five did not come in for their blood draw, and 11 did not show up for their scheduled vaccinations. Fifty-six patients with cancer were consented for the study, and 53 completed all three blood draws and received both shots. Vaccine doses were administered while patients were receiving cytotoxic therapy, and this treatment was not interrupted for vaccine administration. All of the cancer cohort participants received the Pfizer vaccine, BNT162b2; 61 enrolled participants in the control cohort received the Pfizer vaccine, and 12 received the Moderna vaccine, mRNA 1273. For consistency, analyses are restricted to those participants who received the BNT162b2 vaccine. Four control cohort participants were seropositive based on the University of Arizona COVID-19 ELISA pan-Ig Antibody Test11; all of these participants were removed from analyses. The complete study sample size is 53 cancer cohort patients and 50 control cohort participants.
For the interventional booster, all 53 patients in the cancer cohort were considered for continued eligibility and re-consenting, and 38 did not participate. Specific reasons for lack of participation included the following: 11 were ineligible due to going off chemotherapy (11.3%); ten died (unrelated to vaccine as per clinical provider) or were in hospice (19%); six had clinical provider concerns regarding participation (11.3%); and 11 declined to participate (11.3%). The full CONSORT flow diagram is shown in Extended Data Fig. 1. Blood samples were drawn for analysis before the administration of a third booster dose of the Pfizer mRNA vaccine. A final blood draw was performed on all 20 patients between 5 and 11 d from the time of the third vaccine. Enrollment of the first patient was on 8 June 2021, and the final patient was enrolled on 25 June 2021. Patients in this cohort were contacted at 2 weeks (±3 d) and 4 weeks (±7 d) after booster dose for AE and SAE monitoring. All patients with cancer who were included in the observational study (n = 53) were evaluated for inclusion into the interventional study, and there were no interim analyses performed. The per-protocol analyses were to recruit from the 53 evaluable participants from the observational study. This number of participants was unknown at the time of initial IRB evaluation of the protocol, and a placeholder of n = 1,000 was included. The University of Arizona Cancer Center Data Safety and Monitoring Board (DSMB) was the DSMB of record for the interventional component of the trial. The trial was monitored and reviewed for safety quarterly, as it was determined to be a low-risk study per the University of Arizona Cancer Center DSMB charter. The trial protocol is included in the Supplementary Appendix.
PBMC and serum preparation
Twenty milliliters of blood was collected by venipuncture in heparinized Vacutainer tubes (BD), and an additional 10 ml was collected in clot-activating non-heparinized Vacutainer tubes. After more than 30 min at room temperature, non-heparinized tubes were spun at 1,200g for 10 min, and serum was collected and frozen in 1-ml aliquots at −20 °C. For PBMCs, 15 ml of Ficoll-Paque PLUS (Thermo Fisher Scientific) was added to 50-ml Leucosep tubes (Greiner) and spun for 1 min at 1,000g to transfer the density gradient below the filter. Twenty milliliters of blood from the heparinized tubes was then poured into the top of the Leucosep tube and spun at 1,000g for 10 min at room temperature with the brake off. The top plasma layer was carefully collected and frozen at −20 °C, and the remaining supernatant containing PBMCs above the filter was poured into a new 50-ml conical tube containing 10 ml of PBS and spun at 250g for 10 min. Cell pellets were resuspended in RPMI media containing 10% FCS and counted on a Vi-Cell XR (Beckman Coulter). Cells were resuspended to a concentration of 2 × 107 cells per ml in RPMI media containing 10% FCS. An equal volume of 80% FCS + 20% dimethyl sulfoxide was added dropwise and inverted once to mix. Suspensions were distributed at 1 ml per cryovial and frozen overnight at −80 °C in Mr. Frosty freezing chambers (Nalgene). Vials were then transferred to storage in liquid nitrogen.
ELISAs and quantification of antibody titers
Serological assays were performed as previously described. RBD was purchased from GenScript (cat. no. Z03483), and S2 subdomain of the SARS-CoV-2 S glycoprotein was purchased from Sino Biological (cat. no. 40590-V08B). To obtain titers and single-dilution OD450 values, antigens were immobilized on high-adsorbency 384-well plates at 5 ng ml−1. Plates were blocked with 1% non-fat dehydrated milk extract (Santa Cruz Biotechnology, sc-2325) in sterile PBS (Thermo Fisher Scientific HyClone PBS, SH2035) for 1 h, washed with PBS containing 0.05% Tween-20 and overlaid for 60 min with either a single 1:40 dilution or five serial 1:4 dilutions beginning at a 1:80 dilution of serum. Plates were then washed and incubated for 1 h in 1% PBS and milk containing anti-human Pan-Ig HRP-conjugated antibody (Jackson ImmunoResearch, 109-035-064) at a concentration of 1:2,000 for 1 h. Plates were washed with PBS-Tween solution followed by PBS wash. To develop, plates were incubated in tetramethylbenzidine before quenching with 2 N H2SO4. Plates were read for 450-nm absorbance on CLARIOstar Plus from BMG Labtech. All samples were also read at 630 nm to detect any incomplete quenching. Any samples above background 630-nm values were re-run. Area under the curve (AUC) values were calculated in GraphPad Prism (v9).
T cell assays
Frozen PBMCs were thawed by mixing with 10 ml of pre-warmed RPMI 1640 media (Gibco) containing 10% FCS (Peak Serum, PS-FB1), 1× penicillin–streptomycin (HyClone, SV30010) and 0.03 mg ml−1 of DNAse (Sigma-Aldrich, DN25-100) in a 15-ml conical tube and spun at 540g for 5 min. Cell pellets were resuspended in 1 ml of X-VIVO 15 media with gentamicin and phenol red (VWR, 12001-988) containing 5% male human AB serum (Sigma-Aldrich, H4522-100ML) and incubated in 24-well plates overnight at 37 °C with 5% CO2. Next, 250 μl of each sample was plated on a 96-well round-bottom plate and spun at 540g for 3 min and then resuspended in 150 μl of X-VIVO 15 media with 5% male human AB serum containing either 0.6 nmol PepTivator SARS-CoV-2 Prot_S peptide pool (Miltenyi Biotec, 130-126-701) for antigen-specific T cell stimulation or positive control anti-CD3 mAb CD3-2 from Human IFN-γ ELISpot PLUS kit (Mabtech, 3420-4APT-2) or blank media as negative control. In some experiments, 10 μg ml−1 of blocking antibodies against HLA-I (W6/32, BioLegend) and/or HLA-II (Tü39, BioLegend) were included. Antibodies were validated by the manufacturer on human PBMCs. Cell suspensions were transferred to pre-coated IFN-γ ELISpot plates and incubated overnight at 37 °C in 5% CO2. Plates were emptied, washed five times with 200 μl per well of PBS (Thermo Fisher Scientific HyClone PBS, SH2035) and incubated for 2 h at room temperature with 100 μl per well of PBS containing 0.5% FCS and 1 μg ml−1 of detection antibody (7-B6-1-biotin). Plates were washed as above and incubated for 1 h at room temperature with 100 μl per well of PBS-0.5% FCS with 1:1,000 diluted Streptavidin-ALP. Plates were washed as above and developed for 10–15 min with 100 μl per well of substrate solution (BCIP/NBT-plus) until distinct spots emerged. Color development was stopped by washing extensively in tap water and left to dry. Spots were imaged and counted using an ImmunoSpot Versa (Cellular Technology) plate reader.
Virus neutralization assays
All live virus assays were performed at Biosafety Level 3. SARS Coronavirus 2, Isolate USA-WA1/2020 (BEI, NR-52281) was passaged once on Vero (American Type Culture Collection (ATCC), CCL-81) cells at a multiplicity of infection of 0.01 for 48 h. Supernatant and cell lysate were combined, subjected to a single freeze–thaw and then centrifuged at 1,800g for 10 min to remove cell debris. For PRNTs for SARS-CoV-2, Vero cells (ATCC, CCL-81) were plated in 96-well tissue culture plates and grown overnight. Vero cells were confirmed by PCR to be free of mycoplasma. Serial dilutions of serum samples were incubated with 100 plaque-forming units of SARS-CoV-2 for 1 h at 37 °C. Plasma/serum dilutions plus virus were transferred to the cell plates and incubated for 2 h at 37 °C in 5% CO2 and then overlaid with 1% methylcellulose. After 72 h, plates were fixed with 10% neutral buffered formalin for 30 min and stained with 1% crystal violet. Plaques were imaged using an ImmunoSpot Versa plate reader. The most dilute serum concentration that led to ten or fewer plaques was designated as the PRNT90 titer.
Flow cytometry
One milliliter of pre-warmed FCS was added to a frozen cryovial of PBMCs, which was then rapidly thawed in a 37 °C water bath. Samples were poured into 15-ml conical tubes containing 5 ml of pre-warmed RPMI + 10% FCS and spun at 250g for 5 min at room temperature. Supernatants were removed, and pellets were washed once with 500 µl of PBS containing 5% adult bovine serum and 0.1% sodium azide (staining buffer). Cell pellets were then resuspended in 200 μl of staining buffer containing 1 μl each of anti-IgM-FITC (BioLegend, clone MHM-88), anti-IgD-PerCP-Cy5.5 (BioLegend, clone IA6-2), anti-CD11c-Alexa700 (BioLegend, clone Bu15), anti-CD13-PE-Cy7 (BioLegend, clone WM15), anti-CD19-APC-efluor-780 (eBioscience, clone HIB19), anti-CD21-PE-Dazzle (BioLegend, clone Bu32), anti-CD27-BV510 (BioLegend, clone M-T271), anti-CD38-APC (BioLegend, clone HIT2), RBD-PE tetramer and S1-BV421 tetramer. Antibodies were validated by the manufacturer on human PBMCs. Tetramer reagents were assembled by mixing 100 μg ml−1 of C-terminal AviTagged RBD or S1 (ACROBiosystems) with 100 μg ml−1 of streptavidin-PE (eBiosciences) or streptavidin-BV421 (BioLegend), respectively, at a 5:1 molar ratio in which 1/10 of the final volume of streptavidin was added every 5 min. S1 and RBD tetramers were validated by staining Lenti-X 293T cells (Takara Bio) as a negative control or 293T-hACE2-expressing cells (BEI Resources, NR-52511) as a positive control. Lenti-X 293T cells were confirmed to be free of mycoplasma; 293T-hACE2 cells were maintained in media containing 10 μg ml−1 of ciprofloxacin to minimize the chances of mycoplasma contamination. PBMC samples were stained for at least 20 min, washed and filtered through 70-μm nylon mesh. Data were acquired on either a BD LSR2 or a Fortessa flow cytometer. Data were analyzed using FlowJo software.
Statistical methods
The primary statistical endpoint for the observational study was the change in antibody-mediated neutralization of authentic live SARS-CoV-2 PRNT90 titers from baseline to draw 3 between participants in the control cohort and the cancer cohort. This primary endpoint, powered as a non-inferiority hypothesis, was whether vaccine-acquired PRNT90 titers were the same in immunocompromised patients compared to healthy individuals. These methods typically require estimating the outcome under a non-inferiority margin; however, this criterion is not necessary given the obvious superiority of PRNT90 titers seen in healthy participants compared to patients with cancer at draw 3. The primary endpoint for the interventional third vaccine dose study was the paired difference, using a paired t-test statistic, between draw 4 (booster shot) and draw 5 (7 d after booster), with secondary analyses examining the paired difference in RBD titers (as AUC) and spike-specific T cells. Secondary analyses for the observational study included comparing differences in slope and between blood draws—for example, draw 1 to draw 2 and draw 2 to draw 3 between cohorts using repeated-measures analysis of variance (ANOVA) that adjusts for the correlation within an individual by use of an exchangeable covariance matrix and F-test statistics. Pairwise differences between and within cohorts and draw were evaluated using two-way ANOVA and two-sided Sidak-adjusted t-test statistics. Additionally, analysis of covariance was used to evaluate whether age moderated the between-cohort draw 3 differences for the semi-quantitative 1:40 serum dilution ELISA for RBD and S2 spike proteins as well as RBD AUC and neutralizing antibody titers. The cancer cohort was older, on average, with a mean age of 64 years compared to 42 years in the control cohort. The possible mediating effect of age on the immunologic response seen between the cohorts was evaluated in two ways. First, age was added as an interaction effect to draw 3 differences between cohorts, using linear models with age as a continuous variable. Additionally, because the primary age difference between cohorts was the lack of participants in the lowest age quartile (<38 years of age) in the cancer cohort, the draw 3 differences were tested after removing this age group from the control cohort, thus removing the lower age bias that could have been introduced by including these participants; this resulted in a much smaller sample size in the control cohort (n = 23) and was performed using linear modeling with a two-sample, two-sided t-test at the 0.05 level of significance.
For the interventional booster trial, the primary endpoint was the paired change in log10(PRNT90) from the interventional baseline titer (draw 4 since the start of the observational study) to 1 week after the third BNT162b2 dose (draw 5). The primary hypothesis was that there would be an increase in log10(PRNT90) levels between these two blood draws. The test statistic was a two-sided paired t-test. A sample size of at least 35 participants would achieve a power of 0.80 to detect a pairwise difference of 1.17 log10(PRNT90) at a 0.05 level of significance; this sample size assumes a pairwise standard deviation of 2.39, as seen in the observational study. Secondary endpoints would include neutralizing antibodies, RBD titers and T cell ELISpots. The intended sample size was not met for the reasons described in the ‘Participant selection’ section. Thus, the final cohort size for the interventional trial was 20.
All analyses were performed in GraphPad Prism 9 and/or the R programming language (v4.0.5).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
Data are available in the main text, figures, extended data and supplementary files. Raw immune biomarker data are provided in Supplementary Table 2 with individual identifying information removed to preserve patient confidentiality. Flow cytometric files can be requested by contacting D.B. Serum sample requests should be sent to D.B. and will be made available pending sufficient remaining quantities and completion of a material transfer agreement.
References
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020).
Zhou, P. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020).
Polack, F. P. et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Engl. J. Med. 383, 2603–2616 (2020).
Baden, L. R. et al. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 384, 403–416 (2021).
Kuderer, N. M. et al. Clinical impact of COVID-19 on patients with cancer (CCC19): a cohort study. Lancet 395, 1907–1918 (2020).
Herishanu, Y. et al. Efficacy of the BNT162b2 mRNA COVID-19 vaccine in patients with chronic lymphocytic leukemia. Blood 137, 3165–3173 (2021).
Deepak, P. et al. Effect of immunosuppression on the immunogenicity of mRNA vaccines to SARS-CoV-2. Ann. Intern. Med. https://doi.org/10.7326/M21-1757 (2021).
Boyarsky, B. J. et al. Antibody response to 2-dose SARS-CoV-2 mRNA vaccine series in solid organ transplant recipients. JAMA 325, 2204–2206 (2021).
Monin, L. et al. Safety and immunogenicity of one versus two doses of the COVID-19 vaccine BNT162b2 for patients with cancer: interim analysis of a prospective observational study. Lancet Oncol. 22, 765–778 (2021).
Boyarsky, B. J. et al. Immunogenicity of a single dose of SARS-CoV-2 messenger RNA vaccine in solid organ transplant recipients. JAMA 325, 1784–1786 (2021).
Ripperger, T. J. et al. Orthogonal SARS-CoV-2 serological assays enable surveillance of low-prevalence communities and reveal durable humoral immunity. Immunity 53, 925–933 (2020).
Ladner, J. T. et al. Epitope-resolved profiling of the SARS-CoV-2 antibody response identifies cross-reactivity with endemic human coronaviruses. Cell Rep. Med. 2, 100189 (2021).
Anderson, E. M. et al. Seasonal human coronavirus antibodies are boosted upon SARS-CoV-2 infection but not associated with protection. Cell 184, 1858–1864 (2021).
Nguyen-Contant, P. et al. S protein-reactive IgG and memory B cell production after human SARS-CoV-2 infection includes broad reactivity to the S2 subunit. mBio 11, e01991-20 (2020).
Shrock, E. et al. Viral epitope profiling of COVID-19 patients reveals cross-reactivity and correlates of severity. Science 370, eabd4250 (2020).
Ng, K. W. et al. Preexisting and de novo humoral immunity to SARS-CoV-2 in humans. Science 370, 1339–1343 (2020).
Song, G. et al. Cross-reactive serum and memory B-cell responses to spike protein in SARS-CoV-2 and endemic coronavirus infection. Nat. Commun. 12, 2938 (2021).
Hurlburt, N. K. et al. Structural definition of a pan-sarbecovirus neutralizing epitope on the spike S2 subunit. Preprint at https://www.biorxiv.org/content/10.1101/2021.08.02.454829v1 (2021).
Greaney, A. J. et al. Antibodies elicited by mRNA-1273 vaccination bind more broadly to the receptor binding domain than do those from SARS-CoV-2 infection. Sci. Transl. Med. 13, eabi9915 (2021).
Piccoli, L. et al. Mapping neutralizing and immunodominant sites on the SARS-CoV-2 spike receptor-binding domain by structure-guided high-resolution serology. Cell 183, 1024–1042 (2020).
Sekine, T. et al. Robust T cell immunity in convalescent individuals with asymptomatic or mild COVID-19. Cell 183, 158–168 (2020).
Rydyznski Moderbacher, C. et al. Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity. Cell 183, 996–1012 (2020).
Ogbe, A. et al. T cell assays differentiate clinical and subclinical SARS-CoV-2 infections from cross-reactive antiviral responses. Nat. Commun. 12, 2055 (2021).
McMahan, K. et al. Correlates of protection against SARS-CoV-2 in rhesus macaques. Nature 590, 630–634 (2021).
Reynolds, C. J. et al. Discordant neutralizing antibody and T cell responses in asymptomatic and mild SARS-CoV-2 infection. Sci. Immunol. 5, eabf3698 (2020).
Anderson, E. J. et al. Safety and immunogenicity of SARS-CoV-2 mRNA-1273 vaccine in older adults. N. Engl. J. Med. 383, 2427–2438 (2020).
Goel, R. R. et al. Distinct antibody and memory B cell responses in SARS-CoV-2 naïve and recovered individuals following mRNA vaccination. Sci. Immunol. 6, eabi6950 (2021).
McCallum, M. et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2. Cell 184, 2332–2347 (2021).
Cerutti, G. et al. Potent SARS-CoV-2 neutralizing antibodies directed against spike N-terminal domain target a single supersite. Cell Host Microbe 29, 819–833 (2021).
Seifert, M. et al. Functional capacities of human IgM memory B cells in early inflammatory responses and secondary germinal center reactions. Proc. Natl Acad. Sci. USA 112, E546–E555 (2015).
Dogan, I. et al. Multiple layers of B cell memory with different effector functions. Nat. Immunol. 10, 1292–1299 (2009).
Pape, K. A., Taylor, J. J., Maul, R. W., Gearhart, P. J. & Jenkins, M. K. Different B cell populations mediate early and late memory during an endogenous immune response. Science 331, 1203–1207 (2011).
Zuccarino-Catania, G. V. et al. CD80 and PD-L2 define functionally distinct memory B cell subsets that are independent of antibody isotype. Nat. Immunol. 15, 631–637 (2014).
Turner, J. S. et al. Human germinal centres engage memory and naive B cells after influenza vaccination. Nature 586, 127–132 (2020).
Lau, D. et al. Low CD21 expression defines a population of recent germinal center graduates primed for plasma cell differentiation. Sci. Immunol. 2, eaai8153 (2017).
Wong, R. et al. Affinity-restricted memory B cells dominate recall responses to heterologous flaviviruses. Immunity 53, 1078–1094 (2020).
Jenks, S. A. et al. Distinct effector B cells induced by unregulated toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity 49, 725–739 (2018).
Knox, J. J. et al. T-bet+ B cells are induced by human viral infections and dominate the HIV gp140 response. JCI Insight 2, e92943 (2017).
Woodruff, M. C. et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat. Immunol. 21, 1506–1516 (2020).
Kyu, S. Y. et al. Frequencies of human influenza-specific antibody secreting cells or plasmablasts post vaccination from fresh and frozen peripheral blood mononuclear cells. J. Immunol. Methods 340, 42–47 (2009).
Amanna, I. J. Duration of humoral immunity to common viral and vaccine antigens. N. Engl. J. Med. 357, 1903–1915 (2007).
Lavinder, J. J. et al. Identification and characterization of the constituent human serum antibodies elicited by vaccination. Proc. Natl Acad. Sci. USA 111, 2259–2264 (2014).
Purtha, W. E., Tedder, T. F., Johnson, S., Bhattacharya, D. & Diamond, M. S. Memory B cells, but not long-lived plasma cells, possess antigen specificities for viral escape mutants. J. Exp. Med. 208, 2599–2606 (2011).
Smith, K. G. C., Light, A., Nossal, G. J. V. & Tarlinton, D. M. The extent of affinity maturation differs between the memory and antibody-forming cell compartments in the primary immune response. EMBO J. 16, 2996–3006 (1997).
Angyal, A. et al. T-cell and antibody responses to first BNT162b2 vaccine dose in previously SARS-CoV-2-infected and infection-naive UK healthcare workers: a multicentre, prospective, observational cohort study. https://papers.ssrn.com/sol3/papers.cfm?abstract_id=3812375 (2021).
Wumkes, M. L. et al. Serum antibody response to influenza virus vaccination during chemotherapy treatment in adult patients with solid tumours. Vaccine 31, 6177–6184 (2013).
Puthillath, A. et al. Serological immune responses to influenza vaccine in patients with colorectal cancer. Cancer Chemother. Pharmacol. 67, 111–115 (2011).
Abayasingam, A. et al. Long-term persistence of RBD+ memory B cells encoding neutralizing antibodies in SARS-CoV-2 infection. Cell Rep. Med. 2, 100228 (2021).
Mateus, J. et al. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science 370, 89–94 (2020).
Schulien, I. et al. Characterization of pre-existing and induced SARS-CoV-2-specific CD8+ T cells. Nat. Med. 27, 78–85 (2021).
Grifoni, A. et al. Targets of T cell responses to SARS-CoV-2 coronavirus in humans with COVID-19 disease and unexposed individuals. Cell 181, 1489–1501 (2020).
Le Bert, N. et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature 584, 457–462 (2020).
Weiskopf, D. et al. Phenotype and kinetics of SARS-CoV-2–specific T cells in COVID-19 patients with acute respiratory distress syndrome. Sci. Immunol. 5, eabd2071 (2020).
Swadling, L. et al. Pre-existing polymerase-specific T cells expand in abortive seronegative SARS-CoV-2 infection. Preprint at https://www.medrxiv.org/content/10.1101/2021.06.26.21259239v1 (2021).
Zhou, D. et al. Evidence of escape of SARS-CoV-2 variant B.1.351 from natural and vaccine-induced sera. Cell 184, 2348–2361.e6 (2021).
Edara, V.-V. et al. Infection and vaccine-induced neutralizing-antibody responses to the SARS-CoV-2 B.1.617 variants. N. Engl. J. Med. 385, 664–666 (2021).
Werbel, W. A. et al. Safety and immunogenicity of a third dose of SARS-CoV-2 vaccine in solid organ transplant recipients: a case series. Ann. Intern. Med. https://doi.org/10.7326/L21-0282 (2021).
Kamar, N. et al. Three doses of an mRNA Covid-19 vaccine in solid-organ transplant recipients. N. Engl. J. Med. 385, 661–662 (2021).
Khoury, D. S. et al. Neutralizing antibody levels are highly predictive of immune protection from symptomatic SARS-CoV-2 infection. Nat. Med. 27, 1205–1211 (2021).
Gilbert, P. B. et al. Immune correlates analysis of the mRNA-1273 COVID-19 vaccine efficacy trial. Preprint at https://www.medrxiv.org/content/10.1101/2021.08.09.21261290v4 (2021).
Acknowledgements
This work was supported by National Institutes of Health grants R01AI099108 and R01AI129945 (D.B.), R37AG020719 (J.N.Z.) and T32AG058503 (T.J.R.) and by University of Arizona funds (D.B. and R.S.). We thank M. Pepper (University of Washington) and J. Netland (University of Washington) for technical assistance with tetramer stains. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. Schema in Fig. 1a and Extended Data Fig. 9a were created using BioRender.
Author information
Authors and Affiliations
Contributions
Concept and clinical protocol development: R.T.S., P.C., D.P., M.D., B.J.L. and D.B. Experimental design and data analysis: R.W., G.Q., M.S., K.P., T.J.R., J.L.U., M.J., M.W., R.S., J.N.Z., B.J.L. and D.B. Accrual of patients, data entry and adverse event collection: R.T.S., P.C., D.P., S.D., A.W., H.H., A.J.S., A.C. and R.W.
Corresponding authors
Ethics declarations
Competing interests
Sana Biotechnology has licensed intellectual property of D.B. and Washington University in St. Louis. D.B. is a co-founder of Clade Therapeutics. B.J.L. has a financial interest in Cofactor Genomics and Iron Horse Dx. P.C. receives research funding from Pfizer, BioAtla, Zentalis, Genentech, Eli Lilly, Phoenix Molecular Designs, Amgen, Radius Pharmaceuticals, Carrick Therapeutics and Angiochem and served on advisory boards for Novartis, Eli Lilly, Zentalis, AstraZeneca, Amgen, Bayer, Asthenex, Prosigna, Heron, Puma Biotechnology and Oncosec. R.T.S. receives research funding from Merck, Rafael Pharmaceuticals, ImmunoVaccine, Bayer, SeaGen, Exelixis, Pieris, LOXO Oncology, Novocure, NuCana and QED and has served as a consultant/advisor to Merck, Servier, AstraZeneca, EMD Serono, Taiho, QED, Incyte, Genentech and Basilea. The remaining authors declare no competing interests, and no authors are employees of relevant companies.
Additional information
Peer review information Nature Medicine thanks Alexander Huang, Akiko Iwasaki, Naor Bar-Zeev and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Saheli Sadanand and Alison Farrell were the primary editors on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Consort Diagram.
Observational and Interventional (Booster) Studies.
Extended Data Fig. 2 Cellular and serological characterization of blood samples from control and cancer cohorts.
a, PBMC frequencies of blood samples at each timepoint. Two-sided p-values from t-test statistics were calculated for pairwise differences using 2-way ANOVA. Post-hoc testing for multiple comparisons between draws was performed using Sidak’s correction. Comparisons were made between cohorts matched for draw number. All data points represent biological replicates (n = 50 for control cohort; n = 53 for cancer cohort). b, CD19 + B cell frequencies of blood samples at each timepoint. Two-sided p-values from t-test statistics were calculated for pairwise differences using 2-way ANOVA. Post-hoc testing for multiple comparisons between draws was performed using Sidak’s correction. Comparisons were made between cohorts matched for draw number. All data points represent biological replicates (n = 50 for control cohort; n = 53 for cancer cohort). c, CD13 + myeloid cell frequencies of blood samples at each blood draw. Two-sided p-values from t-test statistics were calculated for pairwise differences using 2-way ANOVA. Post-hoc testing for multiple comparisons between draws was performed using Sidak’s correction. Comparisons were made between cohorts matched for draw number. All data points represent biological replicates (n = 50 for control cohort; n = 53 for cancer cohort). d, B cell subset frequencies at each draw, descriptive statistics include mean ± SEM for n = 50 control and n = 53 cancer cohort participants for each subtype. e, Raw ELISA data for quantification of RBD titers shown in Fig. 1d. A serum concentration beginning at 1:80 was serially diluted and area under the curve (AUC) values calculated. Lines connect the same individual at each dilution. Data from the third blood draw are shown for both the control and cancer cohort. Each individual curve represents a biological replicate (n = 50 for control cohort; n = 53 for cancer cohort).
Extended Data Fig. 3 Correlation between RBD-binding antibodies and virus-neutralization.
RBD titers were plotted against PRNT levels for control and cancer cohorts at draws 2 and 3. Pearson correlation analyses were performed. All data points represent biological replicates (n = 50 for control cohort; n = 53 for cancer cohort).
Extended Data Fig. 4 T cell activation in control and cancer cohorts.
a, PBMCs were cultured for 24 h in the presence of an activating anti-CD3 antibody. IFNγ-producing cells were quantified by ELISPOT. Two-sided p-values from t-test statistics were calculated for pairwise differences using 2-way ANOVA. Post-hoc testing for multiple comparisons between draws was performed using Sidak’s correction. Comparisons were made between cohorts matched for draw number. All data points represent biological replicates (n = 50 for control cohort; n = 53 for cancer cohort). b, Spike-specific T cell activation was quantified in the presence or absence of anti-HLA-I and/or anti-HLA-II blocking antibodies. Two-sided p-values from t-test statistics were calculated using 1-way ANOVA. Post-hoc testing for multiple comparisons was performed using Tukey’s correction. P-values >0.05 are not depicted. All data points represent biological replicates (n = 15 for control cohort; n = 16 for cancer cohort).
Extended Data Fig. 5 Age-moderated analysis of immunological parameters.
a, Trajectory between two draws for RBD AUC for each cohort stratified by age quartile. b, Trajectory between two draws for log10(PRNT90) for each cohort stratified by age quartile. c, Trajectory between three draws for Spike-specific T cell frequencies for each cohort stratified by age quartile. RBD AUC and cohort differences were moderated by age (p-value = 0.01). This effect was driven by the effect of age on the control cohort, increasing age was associated with lower RBD AUC, while the cancer cohort levels were similar across the three upper age quartiles. The difference between the cancer cohort and control cohort was different at draw 3 for all age quartiles 2 - 4. Plots show the means and SEM for each draw along with the prediction intervals based on smoothing splines for each cohort There was no statistically significant difference in the relationship between log10(PRNT90) or Spike-specific T-cell frequencies by age. There was a degree of variability in Spike-specific T-cell frequency measurements in both cohorts though the trend in lower draw 3 measures in the cancer cohort remained consistent. The sample size for each age quartile was n = 0, 12, 20, 20 for the cancer cohort and n = 26, 14, 5, 5 for the control cohort.
Extended Data Fig. 6 Immune responses grouped by time post-vaccination or tumor type.
a, RBD-specific antibodies, neutralizing titers, and Spike-specific T cells were plotted as a function of time after the second vaccination. Mean values + SEM are shown. Two-sided p-values from t-test statistics were calculated within each cohort using 1-way ANOVA with post-hoc Tukey’s multiple comparisons test. No significant differences were observed. All data points represent biological replicates (n = 50 for control cohort; n = 53 for cancer cohort). b, RBD-specific antibodies, neutralizing titers, and Spike-specific T cells were plotted as a function of tumor type. Mean values + SEM are shown. Two-sided p-values from t-test statistics were calculated using 1-way ANOVA with post-hoc Tukey’s multiple comparisons test. No significant differences were observed. All data points represent biological replicates. c, RBD-specific antibodies, neutralizing titers, and Spike-specific T cells were plotted comparing participants who either did or did not self-report prior COVID-19. Mean values are shown. All data points represent biological replicates (breast: n = 23; pancreatic cancer: n = 11; biliary: n = 8; colorectal: n = 4; gastroesophogeal: n = 3; ovarian: n = 1; synovial: n = 1).
Extended Data Fig. 7 Quantification of RBD- and S1-specific B cells after vaccination.
RBD- and S1-specific CD19 + B cell frequencies were measured using gating strategies shown in Extended Data Fig. 1 and Fig. 4a. Cells that bind both RBD and S1 are annotated as RBD + , whereas cells that are specific only for S1 are denoted as S1 + . Two-sided p-values from t-test statistics were calculated for pairwise differences using 2-way ANOVA. Post-hoc testing for multiple comparisons between draws was performed using Sidak’s correction. Comparisons were made within cohorts. All data points represent biological replicates (n = 50 for control cohort; n = 53 for cancer cohort).
Extended Data Fig. 8 Quantification of memory B cell subsets after vaccination.
Cells that bind both RBD and S1 are annotated as RBD + , whereas cells that are specific only for S1 are denoted as S1 + . Lines connect the same individual across blood draws, analyses were done on the arcsin of the square-root transformation, to standardize the small percentages. There is a statistically significant difference in slopes between cancer and control cohorts for DN2 RBD + and S1 + (p < 0.0001 and < 0.0001, respectively) and the average rate of change is increasing in the control cohort for both DN2 RBD + and S1 + ; with less dramatic slope increases with DN2 RBD + and a flat trajectory for DN2 S2 + in the cancer cohort over time. Two-sided p-values from t-test statistics were calculated for pairwise differences using 2-way ANOVA. Post-hoc testing for multiple comparisons between draws was performed using Sidak’s correction. Comparisons were made within cohorts or between cohorts by draw, p-values >0.05 are not depicted. All data points represent biological replicates (n = 50 for control cohort; n = 53 for cancer cohort).
Extended Data Fig. 9 Correlation between memory B cells and anamnestic antibody responses.
a, RBD-specific memory B cell frequencies at Draw 3 (calculated as in Fig. 4) were plotted against the difference in RBD antibody titers between Draws 4 and 5. Pearson’s correlation analysis was performed. All data points represent biological replicates (n = 20). b, RBD- and S1-specific memory B cell frequencies at Draw 3 (calculated as in Fig. 4) were plotted against the difference in PRNT-90 titers between Draws 4 and 5. Pearson’s correlation analysis was performed. All data points represent biological replicates (n = 20).
Extended Data Fig. 10 Hierarchical clustering of immunological parameters.
Hierarchical clustering at the variable level, using Spearman’s rank order statistic was performed to evaluate both the correlation (similarity) of immune biomarkers after they are grouped into similar clusters. Of note is the different pattern of both clustering and similarity of the clustered variables between the control and cancer cohorts. Specifically, in the control cohort the B-cell data group into two clusters (including both switched CD21 + and DN2) with a high degree of correlation (spearman correlation of 0.80) a pattern that was not seen in the cancer cohort, within which the only obvious cluster was that of the neutralizing titers, RBD and S2 OD–with a correlation of 0.6.
Supplementary information
Supplementary Information
Supplementary Fig., Supplementary Table 1 and Supplementary Appendix
Supplementary Table 2
Immunological data of all participants.
Rights and permissions
About this article

Cite this article
Shroff, R.T., Chalasani, P., Wei, R. et al. Immune responses to two and three doses of the BNT162b2 mRNA vaccine in adults with solid tumors. Nat Med 27, 2002–2011 (2021). https://doi.org/10.1038/s41591-021-01542-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-021-01542-z
This article is cited by
-
Antibody response to SARS-CoV-2 WT and Omicron BA.4/5 of inactivated COVID-19 vaccine in patients with lung cancer after second and booster immunization
Journal of Hematology & Oncology (2023)
-
Impaired CD4+ T cell response in older adults is associated with reduced immunogenicity and reactogenicity of mRNA COVID-19 vaccination
Nature Aging (2023)
-
Humoral and cellular response to the COVID-19 vaccine in immunocompromised children
Pediatric Research (2023)
-
Immune-related adverse events and disease outcomes after the third dose of SARS-CoV-2 mRNA-BNT162b2 vaccine in cancer patients receiving immune checkpoint inhibitors
Cancer Immunology, Immunotherapy (2023)
-
Longitudinal data on humoral response and neutralizing antibodies against SARS-CoV-2 Omicron BA.1 and subvariants BA.4/5 and BQ.1.1 after COVID-19 vaccination in cancer patients
Journal of Cancer Research and Clinical Oncology (2023)
