
Abstract
Neutrophil extracellular traps (NETs) are chromatin structures loaded with antimicrobial molecules. They can trap and kill various bacterial, fungal and protozoal pathogens, and their release is one of the first lines of defense against pathogens. In vivo , NETs are released during a form of pathogen-induced cell death, which was recently named NETosis. Ex vivo , both dead and viable neutrophils can be stimulated to release NETs composed of either nuclear or mitochondrial chromatin, respectively. In certain pathological conditions, NETs are associated with severe tissue damage or certain auto-immune diseases. This review describes the recent progress made in the identification of the mechanisms involved in NETosis and discusses its interplay with autophagy and apoptosis.
Similar content being viewed by others
Main
Neutrophils have an essential role in innate immunity and are the first cells recruited to the site of infection.1 Human neutrophils are the most abundant leukocytes. They have a very short lifespan, and neutrophil homeostasis is maintained by continuous release of many neutrophils from the bone marrow. Accelerated neutrophil death decreases neutrophil counts (neutropenia) and increases susceptibility to infection. In turn, delayed neutrophil death increases neutrophil counts (neutrophilia) and intensifies innate defenses, possibly promoting chronic inflammation.2 Neutrophils perform their function by engulfing microorganisms or opsonized particles and degrading them by various molecules, and also release lytic enzymes that destroy extracellular pathogens.3 Moreover, they release structures called neutrophil extracellular traps (NETs) that can trap and kill microbes.4
The neutrophil lifespan constitutes a sensitive balance between their function as effecter cells and their potential to inflict tissue damage. In the absence of inflammatory stimuli, neutrophils continuously undergo apoptosis within 24–48 h both in vivo and in cell culture. The large amounts of superoxide produced by the membrane-associated NADPH oxidase in neutrophils have a central role in the destruction of invading pathogens as well as in the resolution of inflammation.5, 6, 7, 8, 9 Congenital defects that prevent NADPH oxidase activity result in chronic granulomatous disease (CGD), which is characterized by exaggerated immune responses10 and recurrent life-threatening infections by a narrow set of microorganisms.11 We recently found that formation of NETs by activated neutrophils requires not only NADPH-oxidase-mediated superoxide production, but also autophagy.12 In this review, we present an overview of the main biochemical and morphological features observed during neutrophil activation and discuss in more detail the contribution of NADPH oxidase, histone citrullination, intracellular calcium levels and autophagy to chromatin decondensation and NET formation.
Release of Extracellular Traps
In 2004, the group of Brinkman and Zychlinsky was the first to report the release of neutrophil extracellular traps (NETs).4 These structures are composed of nuclear chromatin, associated mainly with nuclear histones and many granular antimicrobial proteins (Supplementary Table 1), as well as some cytoplasmic proteins.13 The antimicrobial effects of these NETs, which trap and possibly kill pathogens, are counteracted by DNase treatment or incubation with anti-histone antibodies.4 Submicromolar concentrations of histones and their cleavage products, such as buforin, have potent antimicrobial effects, and their microbicidal effects are enforced when they are concentrated in NET structures.14, 15 NETs are formed in response to a variety of pro-inflammatory stimuli, such as LPS, IL-8 and TNF (Supplementary Table 2), as well as by various microorganisms and pathogens (Supplementary Table 3). Certain pathogens seem to have developed strategies to evade NETs, such as expression of DNases16, 17 or modification of cell wall structures.18 However, release of extracellular chromatin traps is not restricted to neutrophils. Other granular cell types, such as eosinophils19 and mast cells,20 but not basophils, also release extracellular traps (Supplementary Tables 2 and 3). Therefore, the more generalized term ‘ETosis’ was introduced by Wartha et al.21 Typically, mast cell extracellular traps (MCETs) are released in response to stimuli that trigger NET release from neutrophils. MCETs are also composed of nuclear histones and the antimicrobial cathelicidin LL37, as well as tryptase, a granular mast cell marker (Supplementary Table 1).20 Although extracellular trap formation was shown to occur in various clinical conditions (Table 1),22 it remains unclear whether cell death per se is required for the release of NETs in vivo, as we will discuss below.
NET Release by Dying Cells (NETosis)
In the pioneering report on NET release by neutrophils,4 it was proposed that these histone-rich NETs are released from intact viable cells mainly because no cytosolic proteins were detected in NETs, most cells excluded vital dyes, and NETs were detected within 30–60 min after stimulation with triggers known to prolong the neutrophil lifespan, such as IL-8 and LPS. Meanwhile, the same group demonstrated by single cell analysis that NETs arise from a subset of neutrophils undergoing a new form of cell death,23 named NETosis by Steinberg et al.24 Much evidence indicates that NETs are formed in the context of cell death. Indeed, different fungal,25, 26, 27, 28, 29 bacterial4, 16, 17, 23, 26, 30, 31 and protozoal32, 33 strains and species induce the release of NETs (Supplementary Table 3). In contrast to apoptotic cells, NETotic cells do not display eat-me signals such as PS before plasma membrane disruption, preventing their pre-emptive clearance by phagocytes (Figure 1). In contrast to apoptosis or programmed necrosis (necroptosis),34 both the nuclear and granular membranes disintegrate during NETosis, but plasma integrity is maintained.23 This allows the antimicrobial granular cargo to mix with nuclear chromatin.23 No morphologic signs of apoptosis are observed, such as membrane blebbing, nuclear chromatin condensation, PS exposure before plasma membrane rupture and internucleosomal DNA cleavage.23 Caspase activity is only detected during spontaneous neutrophil apoptosis, but not during PMA-induced NETosis.12 Moreover, the kinetics of PMA-induced NETosis are not affected by treatment with the pan-caspase inhibitor zVAD-fmk. This raises the question of whether the caspase-independent cell death can be attributed to necroptosis. However, addition of necrostatin-1 doesn’t affect PMA-induced NETosis.12 These findings indicate that RIP1-mediated necroptosis does not regulate NETosis.

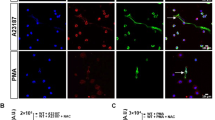
Live cell imaging of subcellular events during ex vivo neutrophil apoptosis and NETosis. (a and c) Cells were incubated with Alexa488-AnnexinV (green) and the cell-impermeable DNA dye propidium iodide (red), (b and d) or with the cell impermeable DNA dye Hoechst (blue), the mitochondrial membrane potential marker TMRM (red) and the cell impermeable DNA dye, propidium iodide (green). Constitutive neutrophil apoptosis is characterized by membrane blebbing, PS exposure (green) and condensation of nuclear chromatin (blue); cells finally undergo secondary necrosis. During induced NETosis, cells display massive vacuolization and decondensation of nuclear chromatin (blue), and PS is not exposed (green) before NET formation (green). Scale bars indicate 10 μm. More information about the visualization of NETs can be found on the following link:89 http://www.jove.com/index/details.stp?ID=1724
Neutrophils stain positive for F-actin after they have undergone (secondary) necrosis, but not after NETosis, and this further shows that NETosis is a cell death modality that differs from apoptosis and necroptosis.30, 31, 35 The presence of histones in NETs further indicates that nuclear—not mitochondrial—chromatin is the major constituent of NETs.
NETosis is activated not only by pathogens and their components, but also by platelets activated with either LPS or the plasma of septic patients also trigger NETosis.36 Anti-neutrophil antibodies that can directly induce NETosis have also been isolated from patients suffering from the auto-immune disease small vessel vasculitis (SVV).37 Anti-neutrophil antibodies are also generated in other diseases, such as SLE and malaria,32, 38 and so they might trigger NETosis in these conditions as well.
A word of caution about the in vivo analysis of NETosis is in order. It was recently reported that fibrin cannot be distinguished from NETs by scanning electron microscopy (SEM), that is, on the basis of morphological criteria.39 Moreover, both NETs and fibrin clots show DNAse-I breakdown, making it unlikely that they can be distinguished in this way.39 In this context, purity of neutrophil preparations is a serious challenge because contamination of neutrophil preparations with platelets can lead to misinterpretation of the process of NETosis. Therefore, in vivo data on NET formation obtained by EM and DNase-I digestion is unlikely to provide unequivocal evidence for NETosis. Consequently, fluorescence microscopy of NET markers (Supplementary Table 1) and immunohistochemical analysis of TEM data have become almost indispensible.39
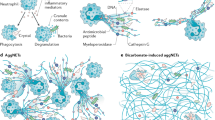
Although the regulation of subcellular events during NETosis remains unclear, increasing evidence indicates that the collapse of the nuclear envelope during NETosis and concurrent intracellular chromatin decondensation are regulated by interplay between histone citrullination, superoxide production and autophagy (Figure 2). We will discuss this in the following section.

Model for the regulation of NETosis. Bacterial DAMPs, such as LPS, are known triggers of autophagy in many cell lines,68 including neutrophils.76 However, purified LPS does not activate NADPH oxidase directly, but only sensitizes for a more powerful NADPH oxidase-derived oxidative burst induced by a subsequent trigger of NADPH oxidase. Formylated peptides are potent triggers of NADPH oxidase. Consequently, fMLP activates NADPH oxidase as well as Histone H3 citrullination. However, fMLP does not induce NETosis. The fMLP induces signaling through Akt/PI3K as well. This cascade PI3K/Akt/mTOR may inhibit autophagy and thereby prevent NETosis. Both ROS production and Histone H3 citrullination are insufficient to mediate the collapse of the nuclear membrane. Accordingly, H2O2 only accelerates neutrophil apoptosis. Induction of autophagy without NADPH oxidase activity, which is observed in CGD neutrophils, also results in enhanced apoptosis with no signs of NETosis. However, ROS (which are sufficient to induce consequent histone H3 citrullination) in combination with induced autophagy leads to chromatin decondensation and collapse of the nuclear membrane and prevents the activity of executioner caspases. This Figure was produced using Servier Medical Art: www.servier.com
Histone citrullination during NETosis
In contrast to apoptosis, NETosis is typically associated with rapid intracellular decondensation of nuclear chromatin.23, 40, 41 This was first observed in promyelocytic HL60 cells differentiated into neutrophil-like cells by stimulation with DMSO41 or retinoic acid.40 Treatment with pro-inflammatory stimuli, such as LPS, IL-8, fMLP or Shigella flexneri induces a marked increase in the citrullination of histone H3.40, 41 Citrullination is the conversion of positively charged arginine side chains into polar but uncharged citrulline side chains, by deimination. Of the five known human peptidylarginine deiminases (PADs) known to catalyze such conversions, PAD4 is the most extensively studied isoform.42 It is expressed by various leukocytes43 including neutrophils44 and immuno-modulatory functions have been ascribed to PAD4 activity.45, 46 Like other PAD isotypes, PAD4 is a Ca2+ dependent enzyme, but only PAD4 possesses a classical nuclear localization signal (NLS).47 Importantly, the citrullination of nuclear histone H3 has been reported to counteract transcription by preventing the methylation of arginine.48 In neutrophils, inhibition of PAD4 prevents citrullination of histone H3 and significantly reduces NET release induced by a Ca2+-ionophore or Shigella flexneri bacteria in differentiated HL60 cells.41 These data suggest that nuclear histones have a regulatory role during NETosis in addition to their direct antimicrobial role. Although NETs released from either differentiated HL60 cells40, 41 or neutrophils40, 49 are positive for citrullinated histone H3, only neutrophils release substantial amounts of NETs. Indeed, no more than 10% of differentiated HL60 cells can generate NETs after stimulation with various triggers of NET release.30, 41, 50 This indicates that histone citrullination is required, but not sufficient to promote NET formation. Although the reason for this difference and the mechanisms regulating NETosis are not clear, these data suggest that additional processes are required to promote NET formation.
Superoxide production during NETosis
Most importantly, when neutrophils are properly activated, they can generate a massive amount of superoxide by activating the NADPH oxidase enzyme complex. ROS were originally reported to be essential for the induction of NETosis,23 but this view has recently been challenged.30 Therefore, the precise role of ROS in NETosis has become a topic of controversy, as we will discuss below. Pharmacological inhibition of NADPH oxidase and interference with the redox metabolism by diphenylene iodonium (DPI) completely prevents PMA-induced NET release.23 In addition, neutrophils from patients with chronic granulomatous disease (CGD), who lack NADPH oxidase activity, are unable to release NETs in response to PMA.23, 30 However, CGD neutrophils regain this ability when incubated with glucose oxidase, which constitutively generates hydrogen peroxide.51 But it should be noted that the glucose oxidase used to induce NETosis in that study was produced in Aspergillus fungi,23 which are known to induce NETosis directly.25, 28, 51, 52 Therefore, we cannot exclude the possibility that PAMPs contaminating the enzyme might have acted as co-stimulatory factors to trigger NETosis.
As mentioned above, NETosis appears as a caspase-independent cell death. One way for ROS to promote this zVAD-fmk-insensitive cell death is by inhibiting caspase activity. Hampton and co-workers have repeatedly shown that ROS generated during an oxidative burst by NADPH oxidase inactivates caspases directly in neutrophils.53, 54, 55 In addition, ROS may indirectly inactivate caspases by NF-κB activation and consequent anti-apoptotic gene expression.9, 56 ROS generated by NADPH oxidase can thus block the apoptotic machinery while activating other processes.
Incubation of neutrophils with hydrogen peroxide results in citrullination of histone H3 by an unknown mechanism.40 Thapsigargin, an inhibitor of SERCA Ca2+ channel, causes an increase in cytosolic Ca2+ levels and was recently shown to trigger NET formation.57 Accordingly, ROS might cause ER and/or mitochondrial damage, resulting in an increase in Ca2+ levels. The increased Ca2+ sustains Ca2+-dependent PAD4 activity, which leads to histone citrullination.40, 41 However, hydrogen peroxide alone is not sufficient for inducing NETosis,12 possibly because of insufficient inhibition of caspase activity or the inability to activate crucial proteases involved in chromatin decondensation during NETosis. NADPH oxidase-derived superoxide mediates the activation of the serine proteases cathepsin G and neutrophil elastase in neutrophils.7 Recently, it was shown that neutrophil elastase contributes to chromatin decondensation by histone cleavage. In contrast, Cathepsin G and proteinase 3 do not contribute to this process.58
Although ROS production apparently promotes NETosis, several lines of evidence indicate the unlikelihood that it is sufficient to trigger it. (1) Mere incubation of neutrophils with low millimolar concentrations of hydrogen peroxide induces only apoptosis, not NETosis.12 (2) Neutrophils isolated from neonates do not generate as much NETs as neutrophils from adults do, despite a similar induction of superoxide production59 (3) Stimulation of neutrophils with fMLP, a potent inducer of NADPH oxidase activity, does not induce NETosis.12, 13 (4) The pro-inflammatory stimuli LPS and IL-8, which were reported to induce NETosis,4, 60 are unable to trigger NADPH oxidase activity, but only sensitize the cells to an oxidative burst.61 This may explain why only some groups observed NET formation in response to LPS and IL-8,4, 60 while others observed a delay in neutrophil apoptosis,2, 62 but no NET formation.12, 36, 63
Serine proteases are also detected in neutrophil-derived NETs (Supplementary Table 1). In addition to the role of neutrophil elastase in NETosis,58 these serine proteases might also act as antimicrobial agents or as modulators of the antimicrobial properties of NET components. In fish and amphibians, exposure of nuclear histones to the proteolytic action of neutrophil granules increases their antimicrobial activity.15 Released histones are also implicated in experimental and human sepsis.64 Extracellular histones, mainly H3 and H4, seem to be biomarkers of disease progression and therapeutic targets in sepsis.64 Of interest, these two histones have much lower affinity than other histones for NET structures,13 which suggests that the H3 and H4 histones dissociate from NETs released by cells. But it is not known to which extent NETosis-related histone modifications, such as histone H3 citrullination, account for changes in the cytotoxicity of histones and their affinity for NETs.36
CGD neutrophils are unable to undergo NETosis in response to the artificial stimulus PMA. This suggests that ROS, derived from NADPH oxidase, are required for the induction of NETosis.23 The inability of CGD neutrophils to undergo NETosis in response to Staphylococcus species further underscored the requirement of ROS herein.23 Similarly, Candida albicans species were shown to induce NETosis only in neutrophils with functional NADPH oxidase activity.26 Moreover, prevention of ROS production abrogated the release of NETs by eosinophils and mast cells.19, 20 Recent evidence, however, demonstrates that NETosis can also occur in a ROS-independent manner.30, 65 For example, stimulation with CXCR2 ligands, but not with PMA, induces NET formation in CGD neutrophils.30 CGD neutrophils are also able to form NETs upon TLR activation (personal communication with D Hartl). Although the mechanisms regulating ROS-independent NETosis are unclear, pharmacological and genetic inhibition of Src kinases can prevent histone citrullination and consequent NET release during CXCR2-dependent NETosis.30
Neutrophil Autophagy
Autophagy refers to self-digesting processes that target intracellular cargo for degradation in endosomes and lysosomes.66 Essential functions of autophagy include the maintenance and homeostatic turnover of organelles and biomolecules, induction of the recycling of organelles and biomolecules during starvation, and the removal of damaged organelles and misfolded proteins during cellular stress.67, 68 Consequently, serious defects in autophagy are lethal.69 Autophagy also contributes to lymphocyte development and survival, antigen presentation and T-cell proliferation. In all these studies, the role of autophagy was studied in vivo by reconstituting liver cells (as a source of hematopoietic precursor cells) from atg5-deficient embryos and neonates into lethally irradiated mice.70, 71, 72 This reconstitution normalizes neutrophil counts, which suggests that autophagy does not mediate neutrophil survival.72
Recent data provide evidence for autophagy's contribution to distinct antimicrobial strategies rather than to neutrophil development or survival. Huang et al.73 demonstrated that autophagy can be induced in murine neutrophils following phagocytosis of either LPS- or IgG-opsonized beads. These results are in line with reports showing that pattern recognition receptors, including Toll-like receptors (TLR) and nucleotide-binding and oligomerization domain (NOD)-like receptors (NLR), induce autophagy in macrophages.74, 75 Mitroulis et al.76 showed that neutrophils too can induce autophagy in response to TLR ligands alone. Consequently, it seems that neutrophil autophagy can be induced in a phagocytosis-dependent or phagocytosis-independent manner.73, 76, 77 The observation that phagocytosis of LPS- or IFN-opsonized particles by neutrophils and macrophages induces autophagy independently of the induced formation of NETs by various microbes suggests that autophagy is an additional mechanism for pathogen clearance.73 This mechanism provides an alternative degradative platform in addition to fusion of phagosomes with lysosomes. In this manner, bacteria that escape phagosomes or prevent phagosome fusion with lysosomes, such as Listeria monocytogenes and Mycobacterium tuberculosis, respectively,78, 79 are confronted by degradative proteases within neutrophils and macrophages made available by autophagy. However, autophagy might also contribute to innate immunity by other mechanisms. In this regard, we recently observed that autophagy is essential for the induction of NETosis.12
Neutrophil Autophagy During NETosis
Fuchs et al.23 originally observed massive vacuolization during the early stages of NETosis. As these vacuoles have a double membrane and are produced before disintegration of the nuclear envelope, they were originally thought to emerge from the nuclear envelope and to promote its collapse before NET formation.23 However, we observed that CGD neutrophils also display massive vacuolization in response to PMA stimulation. Following a period of vacuolization, CGD neutrophils do not seem to undergo NETosis upon activation by PMA, but instead die with hallmarks of apoptosis, such as membrane blebbing, DEVDase activity and, most importantly, apoptotic chromatin condensation instead of the decondensation that precedes NET formation.12 These results suggest that the vesicles do not originate from the nuclear envelope. EM analysis of PMA-stimulated neutrophils clearly revealed autophagosomes in different stages of maturation. Pharmacological inhibition of autophagy did not interfere with the induction of the NADPH oxidase-dependent oxidative burst, but it did interfere with intracellular chromatin decondensation and consequent NETosis, and ultimately resulted in cell death with features of apoptosis.12 This suggests that induction of ROS production is probably insufficient to inhibit caspases, and that the inhibition of caspases requires induction of both autophagy and NADPH oxidase activity. How autophagy contributes to the process of chromatin decondensation remains unclear. The massive extent of vacuolization during NETosis suggests that ER membranes might be assembled as a source of membranes, in addition to possible de novo formation of autophagosomes. If so, a decrease in perinuclear ER membranes could lower morphological constraints on nuclear collapse. In addition, Ca2+ leakage from disrupted ER membranes might also promote PAD4 activity and histone citrullination. However, the prolonged and massive vacuolization in CGD neutrophils did not lead to the collapse of the nuclear envelope, nor did it result in intracellular chromatin decondensation. This argues against a significant contribution of autophagy alone in weakening the nuclear envelope. However, these data cannot rule out that autophagy synergizes with ROS to cause intracellular chromatin decondensation. Further investigation is required to unravel the precise interplay between nuclear collapse on the one hand, and chromatin decondensation on the other hand.
Mitroulis et al.76 recently reported that inflammatory stimuli other than PMA induce neutrophil autophagy. In their study, autophagy was antagonized by antioxidants, suggesting that NADPH oxidase-derived ROS contributes to the induction of autophagy. Although ROS might regulate autophagy,80 they are often insufficient to induce autophagy in many cell lines.81 In neutrophils, stimulation with hydrogen peroxide does not induce autophagy.12 Although superoxide production, when triggered by pathogens and/or a combination of pro-inflammatory stimuli such as PAMPs and cytokines, may contribute to the induction of autophagy during NETosis, our observation of PMA-induced autophagy in CGD neutrophils, which cannot generate superoxide, suggests that superoxide is not required for the induction of autophagy per se.
Stimulation of neutrophils with antibodies against a lectin-like receptor (Siglec-9) induces caspase-independent death of neutrophils associated with formation of autophagosome-like structures.82 Inhibition of ROS production delays both the formation of autophagosome-like structures and the kinetics of cell death.82 Although cell death induced by Siglec-9 was not defined as NETosis, it does demonstrate that the formation of autophagosome-(like) structures might depend on ROS.
Release of NETs by Live Neutrophils
The group of Simon placed extracellular trap formation in a context broader than cell death because they observed that even viable eosinophils and neutrophils release extracellular traps.19, 63 In these studies, eosinophils and neutrophils were pretreated with the pro-inflammatory cytokines IL-5+IFNγ or with GM-CSF+C5a, respectively. Further stimulation of both cell types with LPS resulted in exposure of chromatin exclusively from mitochondria.19, 63 Mitochondrial NETs obviously lack antimicrobial nuclear histones. Nevertheless, antibacterial activity of released mitochondrial chromatin was demonstrated for eosinophil extracellular traps, but not for NETs. The extracellular release of mitochondrial DNA can also activate neutrophils by functioning as a TLR9 ligand,83 while delaying constitutive apoptosis.84 The release of mitochondrial NETs from viable neutrophils is nevertheless very surprising for three reasons: the very small amounts of mitochondria in these cells; the small size of the mitochondrial genome; the absence of antimicrobial histones in such NETs.85, 86
It is noteworthy that neither eosinophils nor neutrophils contain many mitochondria.85 They depend on anaerobic glycolysis to meet their energy requirements,87 which renders them very suitable to operate at sites of inflammation, where oxygen levels are often low. The decrease in mitochondrial activity only seems to affect neutrophil chemotaxis, but not the rate of spontaneous apoptosis.88
Conclusions
As nuclear and mitochondrial NETs are reportedly derived from dying or viable neutrophils, respectively, it is recommendable to differentiate between the two types of NETs by using nuclear markers (Supplementary Table 1). Only a fraction of stimulated neutrophils undergoes NETosis in response to most physiological triggers of NET formation.4, 23 This aspect originally led to the belief that living neutrophils actively release NETs. Time-lapse imaging of single cells has meanwhile revealed that NET formation after various stimuli is most probably associated with cell death, and was therefore named NETosis.24 It is not known why only some neutrophils form NETs. Isolated neutrophils might be more heterogeneous than expected, and it has been proposed that the ability to form NETs is related to the age of the neutrophils.14 Alternatively, there might be inhibitory feedback mechanisms that prevent neighboring cells from sensing NETs in a paracrine way leading to inhibition of NETosis. Released DNA might act as such a sensor.25 All these explanations remain hypothetical.
The recently observed involvement of autophagy in NETosis makes it easier to understand apparently paradoxical data. Differentiated neutrophil-like HL60 cells respond normally in terms of NADPH oxidase activity and consequent histone H3 citrullination to stimuli that induce NET formation. However, only very few NETs, if any, are generated by HL-60 cells, regardless of their differentiation state.30, 41, 50 This may be explained by defects in their ability to induce autophagy. Conversely, LPS induces histone H3 citrullination40 and autophagy76 in mature neutrophils, but NADPH oxidase is normally not activated by LPS alone.61 The induction of NETs by LPS in a DPI sensitive manner suggests that contaminating stimuli are responsible for LPS-induced NADPH oxidase activity and consequent NET formation.59 Although ROS seem necessary for NET formation, they are clearly insufficient to induce NETosis. We therefore hypothesize that the interplay between autophagy, ROS formation and PAD4-dependent histone citrullination promotes the collapse of both nuclear and granular membranes and mediates intracellular chromatin decondensation, while inhibiting the apoptotic machinery. Future research will hopefully elucidate in detail the interplay between these processes and the essential enzymatic activities involved.
Abbreviations
- C5a:
-
complement factor 5a
- CGD:
-
chronic granulomatous disease
- DAMP:
-
damage-associated molecular pattern
- DPI:
-
diphenylene iodonium
- fMLP:
-
N-formylated methionyl-leucyl-phenylalanine
- GM-CSF:
-
granulocyte-macrophage colony-stimulating factor
- IFN:
-
interferon
- IL:
-
Interleukin
- LPS:
-
lipopolysaccharide
- MCET:
-
mast cell extracellular trap
- NET:
-
neutrophil extracellular trap
- Nec-1:
-
necrostatin-1
- NLR:
-
Nod like receptor
- PAD:
-
peptidylarginine deiminase
- PAMP:
-
pathogen associated molecular pattern
- PMA:
-
phorbol-12-myristate-13-acetate
- PS:
-
phosphatidyl serine
- ROS:
-
reactive oxygen species
- SERCA:
-
Sarcoplasmic/Endoplasmic Reticulum Calcium Atpase
- SLE:
-
systemic lupus erythematosus
- SVV:
-
small vessel vasculitis
- TMRM:
-
tetramethyl rhodamine methyl ester
- TNF:
-
tumor necrosis factor
- zVAD-fmk:
-
carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone
References
Nathan C . Neutrophils and immunity: challenges and opportunities. Nat Rev Immunol 2006; 6: 173–182.
Luo HR, Loison F . Constitutive neutrophil apoptosis: mechanisms and regulation. Am J Hematol 2008; 83: 288–295.
Segal AW . How neutrophils kill microbes. Annu Rev Immunol 2005; 23: 197–223.
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS et al. Neutrophil extracellular traps kill bacteria. Science 2004; 303: 1532–1535.
Klebanoff SJ . Myeloperoxidase: contribution to the microbicidal activity of intact leukocytes. Science 1970; 169: 1095–1097.
Meissner F, Seger RA, Moshous D, Fischer A, Reichenbach J, Zychlinsky A . Inflammasome activation in NADPH oxidase defective mononuclear phagocytes from patients with chronic granulomatous disease. Blood 2010; 116: 1570–1573.
Reeves EP, Lu H, Jacobs HL, Messina CG, Bolsover S, Gabella G et al. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 2002; 416: 291–297.
Romani L, Fallarino F, De Luca A, Montagnoli C, D’Angelo C, Zelante T et al. Defective tryptophan catabolism underlies inflammation in mouse chronic granulomatous disease. Nature 2008; 451: 211–215.
Sadikot RT, Zeng H, Yull FE, Li B, Cheng DS, Kernodle DS et al. p47phox deficiency impairs NF-kappa B activation and host defense in Pseudomonas pneumonia. J Immunol 2004; 172: 1801–1808.
Siddiqui S, Anderson VL, Hilligoss DM, Abinun M, Kuijpers TW, Masur H et al. Fulminant mulch pneumonitis: an emergency presentation of chronic granulomatous disease. Clin Infect Dis 2007; 45: 673–681.
van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, Corbeel L et al. Chronic granulomatous disease: the European experience. PLoS ONE 2009; 4: e5234.
Remijsen Q, Vanden Berghe T, Wirawan E, Asselbergh B, Parthoens E, De Rycke R et al. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res 2010; Epub ahead of print 9 November 2010.
Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog 2009; 5: e1000639.
Brinkmann V, Zychlinsky A . Beneficial suicide: why neutrophils die to make NETs. Nat Rev Microbiol 2007; 5: 577–582.
Parseghian MH, Luhrs KA . Beyond the walls of the nucleus: the role of histones in cellular signaling and innate immunity. Biochem Cell Biol 2006; 84: 589–604.
Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B . An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol 2006; 16: 401–407.
Buchanan JT, Simpson AJ, Aziz RK, Liu GY, Kristian SA, Kotb M et al. DNase expression allows the pathogen group A Streptococcus to escape killing in neutrophil extracellular traps. Curr Biol 2006; 16: 396–400.
Wartha F, Beiter K, Albiger B, Fernebro J, Zychlinsky A, Normark S et al. Capsule and D-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell Microbiol 2007; 9: 1162–1171.
Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E et al. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med 2008; 14: 949–953.
Kockritz-Blickwede M, Goldmann O, Thulin P, Heinemann K, Norrby-Teglund A, Rohde M et al. Phagocytosis-independent antimicrobial activity of mast cells by means of extracellular trap formation. Blood 2008; 111: 3070–3080.
Wartha F, Henriques-Normark B . ETosis: a novel cell death pathway. Sci Signal 2008; 1: e25.
Logters T, Margraf S, Altrichter J, Cinatl J, Mitzner S, Windolf J et al. The clinical value of neutrophil extracellular traps. Med Microbiol Immunol 2009; 198: 211–219.
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol 2007; 176: 231–241.
Steinberg BE, Grinstein S . Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci STKE 2007; 2007: e11.
Bruns S, Kniemeyer O, Hasenberg M, Aimanianda V, Nietzsche S, Thywissen A et al. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog 2010; 6: e1000873.
Ermert D, Urban CF, Laube B, Goosmann C, Zychlinsky A, Brinkmann V . Mouse neutrophil extracellular traps in microbial infections. J Innate Immun 2009; 1: 181–193.
Jaillon S, Peri G, Delneste Y, Fremaux I, Doni A, Moalli F et al. The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J Exp Med 2007; 204: 793–804.
McCormick A, Heesemann L, Wagener J, Marcos V, Hartl D, Loeffler J et al. NETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes Infect 2010; 12: 928–936.
Urban CF, Reichard U, Brinkmann V, Zychlinsky A . Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol 2006; 8: 668–676.
Marcos V, Zhou Z, Yildirim AO, Bohla A, Hector A, Vitkov L et al. CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat Med 2010; 16: 1018–1023.
Ramos-Kichik V, Mondragon-Flores R, Mondragon-Castelan M, Gonzalez-Pozos S, Muniz-Hernandez S, Rojas-Espinosa O et al. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis (Edinb) 2009; 89: 29–37.
Baker VS, Imade GE, Molta NB, Tawde P, Pam SD, Obadofin MO et al. Cytokine-associated neutrophil extracellular traps and antinuclear antibodies in Plasmodium falciparum infected children under six years of age. Malar J 2008; 7: 41.
Guimaraes-Costa AB, Nascimento MT, Froment GS, Soares RP, Morgado FN, Conceicao-Silva F et al. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc Natl Acad Sci USA 2009; 106: 6748–6753.
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G . Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010; 11: 700–714.
Palic D, Ostojic J, Andreasen CB, Roth JA . Fish cast NETs: neutrophil extracellular traps are released from fish neutrophils. Dev Comp Immunol 2007; 31: 805–816.
Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med 2007; 13: 463–469.
Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med 2009; 15: 623–625.
Hakkim A, Furnrohr BG, Amann K, Laube B, Abed UA, Brinkmann V et al. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc Natl Acad Sci USA 2010; 107: 9813–9818.
Krautgartner WD, Klappacher M, Hannig M, Obermayer A, Hartl D, Marcos V et al. Fibrin mimics neutrophil extracellular traps in SEM. Ultrastruct Pathol 2010; 34: 226–231.
Neeli I, Khan SN, Radic M . Histone deimination as a response to inflammatory stimuli in neutrophils. J Immunol 2008; 180: 1895–1902.
Wang Y, Li M, Stadler S, Correll S, Li P, Wang D et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol 2009; 184: 205–213.
Anzilotti C, Pratesi F, Tommasi C, Migliorini P . Peptidylarginine deiminase 4 and citrullination in health and disease. Autoimmun Rev 2010; 9: 158–160.
Chang X, Yamada R, Suzuki A, Sawada T, Yoshino S, Tokuhiro S et al. Localization of peptidylarginine deiminase 4 (PADI4) and citrullinated protein in synovial tissue of rheumatoid arthritis. Rheumatology (Oxford) 2005; 44: 40–50.
Asaga H, Nakashima K, Senshu T, Ishigami A, Yamada M . Immunocytochemical localization of peptidylarginine deiminase in human eosinophils and neutrophils. J Leukoc Biol 2001; 70: 46–51.
Proost P, Loos T, Mortier A, Schutyser E, Gouwy M, Noppen S et al. Citrullination of CXCL8 by peptidylarginine deiminase alters receptor usage, prevents proteolysis, and dampens tissue inflammation. J Exp Med 2008; 205: 2085–2097.
Suzuki A, Yamada R, Chang X, Tokuhiro S, Sawada T, Suzuki M et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat Genet 2003; 34: 395–402.
Mastronardi FG, Wood DD, Mei J, Raijmakers R, Tseveleki V, Dosch HM et al. Increased citrullination of histone H3 in multiple sclerosis brain and animal models of demyelination: a role for tumor necrosis factor-induced peptidylarginine deiminase 4 translocation. J Neurosci 2006; 26: 11387–11396.
Cuthbert GL, Daujat S, Snowden AW, Erdjument-Bromage H, Hagiwara T, Yamada M et al. Histone deimination antagonizes arginine methylation. Cell 2004; 118: 545–553.
Marcos V, Nussbaum C, Vitkov L, Hector A, Wiedenbauer EM, Roos D et al. Delayed but functional neutrophil extracellular trap formation in neonates. Blood 2009; 114: 4908–4911.
Papayannopoulos V, Zychlinsky A . NETs: a new strategy for using old weapons. Trends Immunol 2009; 30: 513–521.
Bianchi M, Hakkim A, Brinkmann V, Siler U, Seger RA, Zychlinsky A et al. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 2009; 114: 2619–2622.
Remijsen Q, Vandenabeele P, Willems J, Kuijpers TW . Reconstitution of protection against Aspergillus infection in chronic granulomatous disease (CGD). Blood 2009; 114: 3497.
Fadeel B, Ahlin A, Henter JI, Orrenius S, Hampton MB . Involvement of caspases in neutrophil apoptosis: regulation by reactive oxygen species. Blood 1998; 92: 4808–4818.
Hampton MB, Stamenkovic I, Winterbourn CC . Interaction with substrate sensitises caspase-3 to inactivation by hydrogen peroxide. FEBS Lett 2002; 517: 229–232.
Wilkie RP, Vissers MC, Dragunow M, Hampton MB . A functional NADPH oxidase prevents caspase involvement in the clearance of phagocytic neutrophils. Infect Immun 2007; 75: 3256–3263.
Arruda MA, Rossi AG, de Freitas MS, Barja-Fidalgo C, Graca-Souza AV . Heme inhibits human neutrophil apoptosis: involvement of phosphoinositide 3-kinase, MAPK, and NF-kappaB. J Immunol 2004; 173: 2023–2030.
Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett 2010; 584: 3193–3197.
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A . Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol 2010; 191: 677–691.
Yost CC, Cody MJ, Harris ES, Thornton NL, McInturff AM, Martinez ML et al. Impaired neutrophil extracellular trap (NET) formation: a novel innate immune deficiency of human neonates. Blood 2009; 113: 6419–6427.
Gupta AK, Hasler P, Holzgreve W, Gebhardt S, Hahn S . Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum Immunol 2005; 66: 1146–1154.
Sheppard FR, Kelher MR, Moore EE, McLaughlin NJ, Banerjee A, Silliman CC . Structural organization of the neutrophil NADPH oxidase: phosphorylation and translocation during priming and activation. J Leukoc Biol 2005; 78: 1025–1042.
Francois S, El Benna J, Dang PM, Pedruzzi E, Gougerot-Pocidalo MA, Elbim C . Inhibition of neutrophil apoptosis by TLR agonists in whole blood: involvement of the phosphoinositide 3-kinase/Akt and NF-kappaB signaling pathways, leading to increased levels of Mcl-1, A1, and phosphorylated Bad. J Immunol 2005; 174: 3633–3642.
Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU . Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ 2009; 16: 1438–1444.
Xu J, Zhang X, Pelayo R, Monestier M, Ammollo CT, Semeraro F et al. Extracellular histones are major mediators of death in sepsis. Nat Med 2009; 15: 1318–1321.
Gabriel C, McMaster WR, Girard D, Descoteaux A . Leishmania donovani promastigotes evade the antimicrobial activity of neutrophil extracellular traps. J Immunol 2010; 185: 4319–4327.
Eskelinen EL, Saftig P . Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta 2009; 1793: 664–673.
Eskelinen EL . New insights into the mechanisms of macroautophagy in mammalian cells. Int Rev Cell Mol Biol 2008; 266: 207–247.
Levine B, Kroemer G . Autophagy in the pathogenesis of disease. Cell 2008; 132: 27–42.
Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T et al. The role of autophagy during the early neonatal starvation period. Nature 2004; 432: 1032–1036.
Lee HK, Mattei LM, Steinberg BE, Alberts P, Lee YH, Chervonsky A et al. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 2010; 32: 227–239.
Miller BC, Zhao Z, Stephenson LM, Cadwell K, Pua HH, Lee HK et al. The autophagy gene ATG5 plays an essential role in B lymphocyte development. Autophagy 2008; 4: 309–314.
Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW . A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med 2007; 204: 25–31.
Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA et al. Activation of antibacterial autophagy by NADPH oxidases. Proc Natl Acad Sci USA 2009; 106: 6226–6231.
Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V . Toll-like receptors control autophagy. EMBO J 2008; 27: 1110–1121.
Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, Eissa NT . Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007; 27: 135–144.
Mitroulis I, Kourtzelis I, Kambas K, Rafail S, Chrysanthopoulou A, Speletas M et al. Regulation of the autophagic machinery in human neutrophils. Eur J Immunol 2010; 40: 1461–1472.
Beertsen W, Willenborg M, Everts V, Zirogianni A, Podschun R, Schroder B et al. Impaired phagosomal maturation in neutrophils leads to periodontitis in lysosomal-associated membrane protein-2 knockout mice. J Immunol 2008; 180: 475–482.
Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V . Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004; 119: 753–766.
Uchiya K, Barbieri MA, Funato K, Shah AH, Stahl PD, Groisman EA . A Salmonella virulence protein that inhibits cellular trafficking. EMBO J 1999; 18: 3924–3933.
Chen Y, Azad MB, Gibson SB . Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ 2009; 16: 1040–1052.
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z . Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26: 1749–1760.
von Gunten S, Yousefi S, Seitz M, Jakob SM, Schaffner T, Seger R et al. Siglec-9 transduces apoptotic and nonapoptotic death signals into neutrophils depending on the proinflammatory cytokine environment. Blood 2005; 106: 1423–1431.
Zhang Q, Itagaki K, Hauser CJ . Mitochondrial DNA is released by shock and activates neutrophils via p38 map kinase. Shock 2010; 34: 55–59.
Jozsef L, Khreiss T, Filep JG . CpG motifs in bacterial DNA delay apoptosis of neutrophil granulocytes. FASEB J 2004; 18: 1776–1778.
Maianski NA, Geissler J, Srinivasula SM, Alnemri ES, Roos D, Kuijpers TW . Functional characterization of mitochondria in neutrophils: a role restricted to apoptosis. Cell Death Differ 2004; 11: 143–153.
van Raam BJ, Sluiter W, de Wit E, Roos D, Verhoeven AJ, Kuijpers TW . Mitochondrial membrane potential in human neutrophils is maintained by complex III activity in the absence of supercomplex organisation. PLoS ONE 2008; 3: e2013.
Borregaard N, Herlin T . Energy metabolism of human neutrophils during phagocytosis. J Clin Invest 1982; 70: 550–557.
Fossati G, Moulding DA, Spiller DG, Moots RJ, White MR, Edwards SW . The mitochondrial network of human neutrophils: role in chemotaxis, phagocytosis, respiratory burst activation, and commitment to apoptosis. J Immunol 2003; 170: 1964–1972.
Brinkmann V, Laube B, Abu AU, Goosmann C, Zychlinsky A . Neutrophil extracellular traps: how to generate and visualize them. J Vis Exp 2010; 24: pii: 1724. doi:10.3791/1724.
Vitkov L, Klappacher M, Hannig M, Krautgartner WD . Extracellular neutrophil traps in periodontitis. J Periodontal Res 2009; 44: 664–672.
Lippolis JD, Reinhardt TA, Goff JP, Horst RL . Neutrophil extracellular trap formation by bovine neutrophils is not inhibited by milk. Vet Immunol Immunopathol 2006; 113: 248–255.
Lauth X, Kockritz-Blickwede M, McNamara CW, Myskowski S, Zinkernagel AS, Beall B et al. M1 protein allows Group A streptococcal survival in phagocyte extracellular traps through cathelicidin inhibition. J Innate Immun 2009; 1: 202–214.
Oehmcke S, Morgelin M, Herwald H . Activation of the human contact system on neutrophil extracellular traps. J Innate Immun 2009; 1: 225–230.
Grinberg N, Elazar S, Rosenshine I, Shpigel NY . Beta-hydroxybutyrate abrogates formation of bovine neutrophil extracellular traps and bactericidal activity against mammary pathogenic Escherichia coli. Infect Immun 2008; 76: 2802–2807.
Alghamdi AS, Foster DN . Seminal DNase frees spermatozoa entangled in neutrophil extracellular traps. Biol Reprod 2005; 73: 1174–1181.
Acknowledgements
This research has been supported by Flanders Institute for Biotechnology (VIB), by European grants (FP6 ApopTrain, MRTN-CT-035624; FP7 EC RTD Integrated Project, Apo-Sys, FP7-200767; Euregional PACT II), Belgian grants (Interuniversity Attraction Poles, IAP 6/18) and Flemish grants (Fonds Wetenschappelijke Onderzoek Vlaanderen, G.0875.11 and G.0973.11), Ghent University grants (MRP, GROUP-ID). QR, SL and TVB are postdoctoral researchers with Fonds voor Wetenschappelijk Onderzoek Vlaanderen. PV is holder of a Methusalem grant (BOF09/01M00709) from the Flemish Government. We thank Dr. Amin Bredan (DMBR-VIB, Ghent) for editing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by G Melino
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Remijsen, Q., Kuijpers, T., Wirawan, E. et al. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ 18, 581–588 (2011). https://doi.org/10.1038/cdd.2011.1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2011.1
Keywords
This article is cited by
-
Mechanism and treatment of α-amanitin poisoning
Archives of Toxicology (2023)
-
Neutrophil degranulation and severely impaired extracellular trap formation at the basis of susceptibility to infections of hemodialysis patients
BMC Medicine (2022)
-
Characteristics and Role of Neutrophil Extracellular Traps in Asthma
Inflammation (2022)
-
Circulating cell free DNA response to exhaustive exercise in average trained men with type I diabetes mellitus
Scientific Reports (2021)
-
Cell death in chronic inflammation: breaking the cycle to treat rheumatic disease
Nature Reviews Rheumatology (2020)
