
Abstract
Background
Adverse drug reactions (ADRs) of biopharmaceuticals can be batch or product specific, resulting from small differences in the manufacturing process. Detailed exposure information should be readily available in systems for postmarketing safety surveillance of biopharmaceuticals, including spontaneous reporting systems (SRSs), in which reports of ADRs are collected.
Objective
The aim of this study was to explore the current status of traceability of biopharmaceuticals in the US and the EU up to patient level in SRSs.
Design and Setting
A cross-sectional study was conducted over the period 2004–2010, including ADR reports from two major SRSs: the FDA Adverse Event Reporting System (FAERS) in the US and EudraVigilance (EV) in the EU.
Main Outcome Measures
The availability of batch numbers was determined for biopharmaceuticals, and compared with small molecule drugs. For biopharmaceuticals for which a biosimilar has been approved for marketing in the EU, the identifiability of the product (i.e. the possibility of distinguishing the biosimilar from the reference biopharmaceutical) was determined.
Results
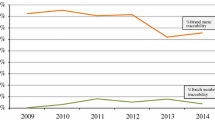
A total of 2,028,600 unique ADR reports were identified in the FAERS, reporting a total of 591,380 biopharmaceuticals (of which 487,065 were suspected). In EV there were 2,108,742 unique ADR reports, reporting a total of 439,971 biopharmaceuticals (356,293 suspected). Overall, for 24.0 % of the suspected biopharmaceuticals in the FAERS and 7.4 % of the suspected small molecule drugs (p < 0.001) batch numbers were available. A similar pattern was seen in EV: for 21.1 % of the suspected biopharmaceuticals batch numbers were available, compared with only 3.6 % of the small molecule drugs (p < 0.001). In both SRSs, consumers were most likely to report a batch number for suspected biologicals (36.3 % in the FAERS and 40.7 % in EV). A total of 13,790 biopharmaceuticals (9,759 suspected) for which a biosimilar has been approved in the EU were identified in EV. For 90.4 % of these biopharmaceuticals and 96.2 % of the suspected biopharmaceuticals the product was clearly identifiable.
Conclusion
This study underlines the need for improving traceability of biopharmaceuticals, in particular with respect to individual batches, allowing better identification and monitoring of postmarketing safety issues related to biopharmaceuticals.




Similar content being viewed by others


References
Giezen TJ, Mantel-Teeuwisse AK, Leufkens HG. Pharmacovigilance of biopharmaceuticals: challenges remain. Drug Saf. 2009;32(10):811–7.
Declerck PJ. Biotherapeutics in the era of biosimilars: what really matters is patient safety. Drug Saf. 2007;30(12):1087–92.
Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian JJ, Martin-Dupont P, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346(7):469–75.
Ebbers HC, Mantel-Teeuwisse AK, Moors EH, Schellekens H, Leufkens HG. Today’s challenges in pharmacovigilance: what can we learn from epoetins? Drug Saf. 2011;34(4):273–87.
Chirino AJ, Mire-Sluis A. Characterizing biological products and assessing comparability following manufacturing changes. Nat Biotechnol. 2004;22(11):1383–91.
Sharma B. Immunogenicity of therapeutic proteins. Part 3: impact of manufacturing changes. Biotechnol Adv. 2007;25(3):325–31.
European Medicines Agency. Note for guidance on biotechnological/biological products subject to changes in their manufacturing process (CPMP/ICH/5271/03). June 2005. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500002805.pdf. Accessed 7 May 2013.
US FDA. Demonstration of comparability of human biological products, including therapeutic biotechnology-derived products. April 1996. http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm122879.htm. Accessed 7 May 2013.
Schellekens H. When biotech proteins go off-patent. Trends Biotechnol. 2004;22(8):406–10.
Schiestl M, Stangler T, Torella C, Cepeljnik T, Toll H, Grau R. Acceptable changes in quality attributes of glycosylated biopharmaceuticals. Nat Biotechnol. 2011;29(4):310–2.
Buttel IC, Chamberlain P, Chowers Y, Ehmann F, Greinacher A, Jefferis R, et al. Taking immunogenicity assessment of therapeutic proteins to the next level. Biologicals. 2011;39(2):100–9.
Mellstedt H, Niederwieser D, Ludwig H. The challenge of biosimilars. Ann Oncol. 2008;19(3):411–9.
Schellekens H. Bioequivalence and the immunogenicity of biopharmaceuticals. Nat Rev Drug Discov. 2002;1(6):457–62.
Walsh G. Biopharmaceutical benchmarks 2010. Nat Biotechnol. 2010;28(9):917–24.
Wadman M. Bills target biosimilar drugs. Nature. 2009;458:394–5.
McCamish M, Woollett G. Worldwide experience with biosimilar development. MAbs. 2011;3(2):209–17.
European Medicines Agency. Note for guidance—EudraVigilance human—processing of safety messages and individual case safety reports (ICSRs). 15 October 2010. http://eudravigilance.ema.europa.eu/human/docs/guid%C2%AFP%C2%AFTechnical%20Documentation%C2%AFEMEA-H-20665-04-en-Final_Revision_2.pdf. Accessed 7 May 2013.
US FDA. Postmarketing reporting of adverse experiences. Code of Federal Regulations Title 21 (21 CFR 600.80). Silver Spring: US FDA; 2012.
US FDA. Postmarketing reporting of adverse drug experiences, Code of Federal Regulation Title 21, (21 CFR 314.80). Silver Spring: US FDA; 2012.
Regulation (EC) No 726/2004 of The European Parliament and of the Council of 31 March 2004, laying down Community procedures for the authorisation and supervision of medicinal products for human and veterinary use and establishing a European Medicines Agency. http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2004:136:0001:0033:en:PDF. Accessed 7 May 2013.
Crommelin DJA, Storm G, Verrijk R, de Leede L, Jiskoot W, Hennink WE. Shifting paradigms: biopharmaceuticals versus low molecular weight drugs. Int J Pharm. 2003;266(1–2):3–16.
Vonberg RP, Gastmeier P. Hospital-acquired infections related to contaminated substances. J Hosp Infect. 2007;65(1):15–23.
Thorpe SJ, Fox BJ, Dolman CD, Lawrence J, Thorpe R. Batches of intravenous immunoglobulin associated with adverse reactions in recipients contain atypically high anti-Rh D activity. Vox Sang. 2003;85(2):80–4.
Kurz X, Domergue F, Slattery J, Segec A, Szmigiel A, Hidalgo-Simon A. Safety monitoring of Influenza A/H1N1 pandemic vaccines in EudraVigilance. Vaccine. 2011;29(26):4378–87.
de Bousingen DD. Former French ministers on trial for infected blood-products scandal. Lancet. 1999;353(9153):653.
Esmonde TF, Will RG, Slattery JM, Knight R, Harries-Jones R, de Silva R, et al. Creutzfeldt-Jakob disease and blood transfusion. Lancet. 1993;341(8839):205–7.
Eaton L. Haemophilia patient had variant CJD agent in spleen. BMJ. 2009;18(338):b705.
van Hunsel F, Harmark L, Pal S, Olsson S, van Grootheest K. Experiences with adverse drug reaction reporting by patients: an 11-country survey. Drug Saf. 2012;35(1):45–60.
Zuñiga L, Calvo B. Biosimilars: pharmacovigilance and risk management. Pharmacoepidemiol Drug Saf. 2010;19(7):661–9.
Fox JL. Debate over details of US biosimilar pathway continues to rage. Nat Biotechnol. 2012;30(7):577.
Lovis C. Traceability in healthcare: crossing boundaries. Yearb Med Inform. 2008;47(Suppl 1):105–13.
O’Dowd A. UK launches inquiry into safety of PIP breast implants. BMJ. 2012;3(344):e11.
Petitjean S. Commission to enhance traceability of medical devices. http://www.europolitics.info/commission-to-enhance-traceability-of-medical-devices-art324118.html. Accessed 22 Mar 2012.
European Federation of Pharmaceutical Industries and Associations. EFPIA product verification project. April 2011. http://www.efpia.eu. Accessed 16 Jan 2011.
Directive 2010/84/EU of The European Parliament and of the Council of 15 December 2010, amending, as regards pharmacovigilance, Directive 2001/83/EC on the Community code relating to medicinal products for human use. http://eudravigilance.ema.europa.eu/human/docs/Directives/dir_2010_84_en.pdf. Accessed 7 May 2013.
Hauben M, Reich L, DeMicco J, Kim K. ‘Extreme duplication’ in the US FDA Adverse Events Reporting System database. Drug Saf. 2007;30(6):551–4.
Acknowledgments
The Department of Pharmacoepidemiology and Clinical Pharmacology, Utrecht Institute for Pharmaceutical Sciences, has received unrestricted research funding from the Netherlands Organisation for Health Research and Development (ZonMW), the Dutch Health Care Insurance Board (CVZ), the Royal Dutch Pharmacists Association (KNMP), the private-public funded Top Institute Pharma (www.tipharma.nl, includes co-funding from universities, government and industry), the EU Innovative Medicines Initiative (IMI), EU 7th Framework Program (FP7), the Dutch Medicines Evaluation Board and the Dutch Ministry of Health and Industry (including GlaxoSmithKline, Pfizer and others). The views expressed in this article are the personal views of the author(s) and may not be understood or quoted as being made on behalf of or reflecting the position of the EMA or one of its committees or working parties.
Conflicts of interest
Niels S. Vermeer, Sabine M.J.M. Straus, Aukje K. Mantel-Teeuwisse, Francois Domergue, Toine C.G. Egberts, Hubert G.M. Leufkens and Marie L. De Bruin have no conflicts of interest that are directly relevant to the content of this review.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below are the links to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Vermeer, N.S., Straus, S.M.J.M., Mantel-Teeuwisse, A.K. et al. Traceability of Biopharmaceuticals in Spontaneous Reporting Systems: A Cross-Sectional Study in the FDA Adverse Event Reporting System (FAERS) and EudraVigilance Databases. Drug Saf 36, 617–625 (2013). https://doi.org/10.1007/s40264-013-0073-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-013-0073-3