Article Text

Abstract
Objectives To provide an update of the EULAR rheumatoid arthritis (RA) management recommendations addressing the most recent developments in the field.
Methods An international task force was formed and solicited three systematic literature research activities on safety and efficacy of disease-modifying antirheumatic drugs (DMARDs) and glucocorticoids (GCs). The new evidence was discussed in light of the last update from 2019. A predefined voting process was applied to each overarching principle and recommendation. Levels of evidence and strengths of recommendation were assigned to and participants finally voted on the level of agreement with each item.
Results The task force agreed on 5 overarching principles and 11 recommendations concerning use of conventional synthetic (cs) DMARDs (methotrexate (MTX), leflunomide, sulfasalazine); GCs; biological (b) DMARDs (tumour necrosis factor inhibitors (adalimumab, certolizumab pegol, etanercept, golimumab, infliximab including biosimilars), abatacept, rituximab, tocilizumab, sarilumab and targeted synthetic (ts) DMARDs, namely the Janus kinase inhibitors tofacitinib, baricitinib, filgotinib, upadacitinib. Guidance on monotherapy, combination therapy, treatment strategies (treat-to-target) and tapering in sustained clinical remission is provided. Safety aspects, including risk of major cardiovascular events (MACEs) and malignancies, costs and sequencing of b/tsDMARDs were all considered. Initially, MTX plus GCs is recommended and on insufficient response to this therapy within 3–6 months, treatment should be based on stratification according to risk factors; With poor prognostic factors (presence of autoantibodies, high disease activity, early erosions or failure of two csDMARDs), any bDMARD should be added to the csDMARD; after careful consideration of risks of MACEs, malignancies and/or thromboembolic events tsDMARDs may also be considered in this phase. If the first bDMARD (or tsDMARD) fails, any other bDMARD (from another or the same class) or tsDMARD (considering risks) is recommended. With sustained remission, DMARDs may be tapered but should not be stopped. Levels of evidence and levels of agreement were high for most recommendations.
Conclusions These updated EULAR recommendations provide consensus on RA management including safety, effectiveness and cost.
- Arthritis, Rheumatoid
- Antirheumatic Agents
- Biological Therapy
Statistics from Altmetric.com
In 2010, the EULAR has developed recommendations for the management of rheumatoid arthritis (RA) with disease-modifying antirheumatic drugs (DMARDs).1 Thereafter, updates of these recommendations have been produced every 3 years, as insights have evolved, and new classification criteria,2 new definitions of remission,3 new treatment strategies4 and many new drugs have emerged. The last update of the recommendations was in 2019.5
While updates of recommendations are neither automatic nor mandatory, they become a necessity if new information arises that requires consideration of its potential impact on an existing guidance document. There is good reason to end the discussion sections of all previous publications on the EULAR RA management recommendations with a sentence like: ‘With the current rate of development, we expect an update of these recommendations to be necessary in about 3–4 years’.5 The convenor of the Task Force, the methodologist and its other members are required to note carefully developments in the field and evaluate if they are important enough to propose an update to the EULAR Council.
When reviewing the developments in the field, many questions arise, such as: (1) which drugs have recently been approved or have completed successful phase 3 trials? (2) Have any previously unrecognised safety concerns become apparent from clinical trials or real-world data analyses? (3) What new information has arisen from the patient perspective or strategic trials? (4) Has the level of evidence (LoE) increased for recommendations previously based on relatively low evidence? (5) Have any of the previous recommendations been contradicted by new data? and (6) Have new data been published on questions raised in previous research agendas?
Two circumstances made it particularly advisable to revisit the current recommendations. First, in 2021, the US Food and Drug Administration (FDA) released a document and warning on cardiovascular and malignancy risks of tofacitinib in comparison with TNF-inhibitors, based on analyses of a randomised trial.6 This was followed by the publication of the full paper in early 2022.7 Second, in the most recent update of the RA management guidelines of the American College of Rheumatology (ACR), the use of glucocorticoids (GCs) was distinctly discouraged, even though the evidence level for this new guideline was low to moderate, reasoning that the toxicity of GCs outweighs the benefits.8 Given that EULAR in its recommendations hitherto has strongly advocated the use of short-term GCs as a bridging therapy when starting conventional synthetic (cs) DMARD therapy, with subsequent rapid tapering of GCs to discontinuation,5 revisiting this issue was warranted.
Management recommendations should provide some guidance on what experts consider is a rational, or maybe even the most effective approach to treating a disease, especially when so many drugs are available as is now the case for RA. What is the therapeutic goal and how should it be targeted? Which medicines should be used in newly diagnosed patients? What should be the sequence if an initial or a subsequently applied drug fails to lead to the therapeutic goal due to lack of efficacy or adverse events? These and more questions must be answered in the context of such guidance and the input from patients and experts with different areas of expertise is important for its generation. In addition, costs must be accounted for, not only in less affluent countries, but also in those in which medicines are affordable but the healthcare system requires to limit expenditures. All of this is part and parcel of the EULAR RA management recommendations and will be included in the current update.
Methods
Steering committee and task force
In line with the EULAR standard operating procedures (SOPs) for developing recommendations9 and the AGREE II document,10 this update started with the approval of the proposal by the EULAR Council. Subsequently, the convenor (JSS) and the methodologist (RBML) invited several experts to serve on the steering committee and others to participate in the expanded task force. The experts were mostly rheumatologists and included patient research partners and non-physician health professionals. The steering committee (JSS, RBML, DA, RC, CJE, JEP, DvdH, TT, PV, KLW, SdS, TAS, SAB, AK and AS) included the three systematic literature research (SLR) researchers (SAB, AK, AS), a non-medical health professional (TAS) and a patient research partner (SdS).
The 2022 update of the EULAR RA management recommendations occurred during the COVID-19 pandemic and travel restrictions prevented some originally invited task force members from attending the meeting in person. Virtual attendance in the form of a hybrid meeting was enabled for overseas participants, so that these members could follow the discussions and raise their voices at any point in time. Nevertheless, several overseas members came to the meeting in Zurich in person, and all European members were requested to attend the face-to-face meeting to facilitate efficient development of the recommendations.
The total task force consisted of the steering group of 15 individuals and an additional 33 experts. Non-European participants came from Africa (EvD), Asia (ZL and TT), Australia (PN), Latin America (MHC and EM) and North America (JMB, JEP and KLW). The participation of non-European experts ensured that the recommendations would also receive input from specialists who practice in other regions of the world, allowing incorporation of a global perspective as well as information and suggestions from low-income countries. EMEUNET, the EULAR network of young rheumatologists, was also represented (KL and FR). Most people attended the face-to-face meeting in Zurich in April 2022; some of the international participants could only join virtually but were present for all or most of the session.
Most participants were invited based on their expertise in the field; four members (KEA, KC, GR and NS) were selected after an open call by EULAR based on their interest and motivations. Of note, some people who originally had agreed to attend, cancelled unexpectedly, among them another patient research partner; a third one, who originally had agreed to be part of the task force, was among those invitees who could not attend at that date. The full task force also included two additional non-medical health professional (KEA and TPMVV). All taskforce members were experienced in the treatment of RA. One member was an infectious disease and epidemiology specialist (KLW) but also had experience related to rheumatic diseases and their treatment.
The process started with a virtual meeting of the steering committee in October 2021 to define the scope of the activity and especially the research questions for the three preparatory SLR activities. Subsequently, the SLRs were performed, this time not only focusing on the topics of (1) safety and (2) efficacy of cs, biological (b) and targeted synthetic (ts) DMARDs, but (3) additionally also on GCs. For the first two SLRs, the activities carried out for the 2019 update served as a starting point11 12 and focused on reviewing publications since then. In contrast, an SLR dedicated to GCs was previously only performed in 201013 and, therefore, this SLR on GCs had to encompass a much longer period of time. Information on pragmatic strategy trials was also included in the efficacy and GC SLRs. The SLR results, whose details are published separately,14–16 were presented to the steering committee in full detail and to the whole task force in an abbreviated fashion focusing on the most important findings. The steering committee thoroughly discussed the SLR results and formulated suggestions for an update of the recommendations which were then presented to the whole task force and discussed in greater detail. To facilitate discussions, the task force was split into three subgroups, each charged to address specific recommendations pertaining to the topics of the individual SLRs (efficacy of b/tsDMARDs, safety of b/tsDMARDs and efficacy and safety of GCs). Thereafter, the subgroups reported back to the whole group, presented the results of their discussions, including new proposals for recommendation wordings for further amendment and voting.
Of note, all task force members declared their conflicts of interest before the meeting to the EULAR Council. Considering one of the major points of the discussions, namely the benefits and risks of Janus kinase inhibitors (JAKi), it must be borne in mind that many of the rheumatologists in the task force had been involved with clinical trials of these agents and/or in advisory board meetings of companies producing these drugs. Therefore, they may have had longer experience with JAKi’s and more detailed information than those not involved in such activities. While none of them declared that this would construe a conflict, inadvertent conflicts may reside and this is mentioned upfront for reasons of transparency and in addition to the declaration of interests at the end of the publication. Most of them also participated in trials and/or advisory boards for companies producing other agents, including bDMARDs.
Consensus building
A few procedural directions were in place for the process of the consensus building activity. First, focus was directed at changing only those recommendations for which new evidence demanded such a change and to try refraining from making minor amendments (ie, changing a word for semantic reasons or changing the position of a word), unless such modification helped mitigate potential misunderstanding. Second, as per previous agreement when developing these recommendations, not-yet-approved drugs with evidence from phase 3 clinical trials available could be considered in the recommendations to anticipate imminent future developments. It is evident that such drugs may only be prescribed after approval by regulatory agencies. However, no such drug was discussed this time. Aside from the data presented in the safety SLR,15 physicians should always also gain information from the summary of product characteristics or label to be fully informed about risks and other safety aspects, which are not discussed in this paper.
After the presentation of the SLR results and the proposals of the steering committee and the breakout groups, the task force further evaluated the new evidence. The voting on keeping recommendations, amending them, deleting old or adding new recommendations took place with the requirement of at least a 75% majority in favour of keeping old versions or any new formulation (or other changes); if that threshold was not reached, the discussion went on and the wording of a particular recommendation was modified, thereafter requiring more than two-thirds (67%) of the votes; if that failed to be reached, a further amendment was made and at that stage more than 50% of the votes were required or else the proposal was rejected.
The task force continued to use the Oxford Centre for Evidence-Based Medicine LoE approach rather than other more formalistic systems, such as the Grading of Recommendations, Assessment, Development and Evaluation (GRADE) system, because ‘what GRADE has gained in accuracy, it may have lost in simplicity and efficiency.’17 Moreover, ‘busy clinicians, who only have a few minutes to answer a clinical question, will need a fast and frugal ‘heuristic search’ tool to find and use the likely best evidence’17 and thus practical applicability of the conclusions. In line with the EULAR SOPs it was, indeed, felt that recommendations for the management of complex diseases such as RA should be based on all available evidence and formulated by an expert committee in a way that makes them easy to understand and follow, and also allows the development of a clear and succinct graphic algorithm.
After the meeting, the results were summarised and the wording voted on in the form of a table with the respective LoEs and strengths of recommendation (SoRs). This table was sent to the task force members for anonymised voting on the levels of agreement (LoAs) with each overarching principle and recommendation, using a scale of 0–10 (0 indicating no agreement at all and 10 indicating full agreement). Aside from the LoEs and SoRs, the mean LoAs as well as the percentage of votes of 8 or more will be presented for each item.
Once the manuscript summarising the updated recommendations and the results of the discussion was finalised, it was sent to the steering group for comments and suggestions for change. Once these were incorporated, a second round of manuscript assessment by the whole task force took place. After the respective adaptations were made, the manuscript was sent first to the EULAR Council and after its approval the final agreed version submitted for publication, together with the three SLR papers.
Results
The results of the SLRs will not be presented here in detail but are presented in respective parallel publications.14–16 However, if pertinent for the explanation of the results, parts of these data will be mentioned.
The glossary previously developed5 will be used here in an amended form for clarity and ease of following the recommendations (table 1).
Glossary and definitions (after76)
It is noteworthy, that since the last update, no new drug class has been approved. Two newer JAKi, upadacitinib and filgotinib, were licensed since then in the European Union and other parts of the world, but based on phase 3 trials they had already been addressed in the 2019 update. Consequently, the focus of the task force was on safety aspects of JAKi and the use of GCs in the sense of the strategic use of available agents and their preferred order.
Overarching principles
As in previous versions of these recommendations, the task force continued to use overarching principles for information on the general aspects of the management of RA that relate to common sense and need no specific evidence levels, but it remained important to reiterate them as the foundation of all treatment approaches.
Treatment of patients with RA should aim at the best care and must be based on a shared decision between the patient and the rheumatologist. This principle remained unchanged both in its wording and its place as item A. However, the patient research partner suggested to clearly mention in the accompanying text that ‘shared decision’ implied the recognition of patient preferences, to which all participants agreed. Otherwise, there was no further discussion on this point and 100% of the participants voted to keep the wording as it was. LoA was 10.0±0.
Treatment decisions are based on disease activity, safety issues and other patient factors, such as comorbidities and progression of structural damage. The task force did not see any reason for a change but it was discussed to specifically mention the importance of thorough history taking and information provided by the patient; 100% of task force members voted to keep this principle as is; LoA was 9.9±0.4.
Rheumatologists are the specialists who should primarily care for patients with RA. This principle has evoked debates, as in previous task forces–it was suggested that the word ‘primarily’ should be deleted. Still, as argued before, in many countries of the world either rheumatology training or rheumatologists are not available at all, or the number of trained rheumatologists is not high enough to take care of all patients with RA. It was finally decided to uphold the original wording and word order, with a supporting vote of 97.8% and an LoA of 9.8±0.9.
Patients require access to multiple drugs with different modes of action to address the heterogeneity of RA; they may require multiple successive therapies throughout life. This item first entered the principles in 2019 and was fully endorsed in 2022. However, it was suggested to mention in its context that cycling should not occur too rapidly, since all agents may need several weeks or months to develop their full effects and, therefore, efficacy of every new treatment should not be judged earlier. To this end, the general treat-to-target approach, as recommended in the EULAR guidance, focuses on at least a 50% improvement in disease activity within 3 months and the attainment of the main treatment target, which is remission in early and low disease activity in long-standing disease, at about 6 months. Hence, drug-cycling before these benchmarks and in the context of incidental and transient flares should be avoided, unless safety mandates a change. The voting arrived at 100% agreement, and the LoA at 9.8±0.6.
RA incurs high individual, medical and societal costs, all of which should be considered in its management by the treating rheumatologist. Again, all task force members agreed with this item (100% of the votes) which as a general principle also puts forward the EULAR Task Force’s view on costs: if two drugs are equally appropriate for a specific patient, then the drug that is less costly should be used. This adage starts with oral versus parenteral methotrexate (MTX), expands to the choice of biosimilar (bs) DMARDs versus biologic originator (bo) DMARDs, and ends at comparisons among bDMARDs and tsDMARDs. Costs, however, have to be seen from a general perspective, as they do not only relate to the price of a drug in the pharmacy, but also to hospital or societal costs and out-of-pocket expenses for the patients. It should also be borne in mind that EULAR endorsed the use of biosimilar (bs) DMARDs for this reason almost a decade ago, long before other organisations have done so.18 The use of bsDMARDs has already helped to reduce drug-costs substantially, and a rational, evidence-based treatment prescription policy further helps to reduce costs: if two drugs are similarly effective and safe for an individual patient, the less expensive one should be used—a very standard attitude in all areas of medicine.19 Of particular note, healthcare systems (and with them patients and rheumatologists) in resource-poor countries are severely resource-constrained in terms of finances and human resources, and this also pertains to some high income countries, especially in the field of rheumatology. However, it is important that not only rheumatologists but also payers follow evidence in medicine; since rheumatologists are central as patient advocates, it is important that they present the wealth of available data to the funders and all other health professionals involved with RA management, as without this funding for advanced technologies treatment success may less likely be achieved. Treatment of RA is not just about costly treatment options, such as targeted DMARDs, but includes correct treatment from the outset of the patient’s journey as described in these recommendations. All members agreed with this principle at the meeting and the LoA was 9.7±0.6.
Individual recommendations
The task force’s deliberations resulted in 11 recommendations, 1 less than in 20195 and 4 less than in the first version in 2010.1 For most of the recommendations the LoE was high and this is shown in table 2. Recommendations 1–5 as well as 7 and 9 remained unchanged and recommendations 11 and 12 from 2019 were brought together as recommendation 11. Although seven recommendations remained unchanged, we will provide a summary of the debates around them in the following section.
EULAR RA management recommendations—2022 update
Therapy with DMARDs should be started as soon as the diagnosis of RA is made. In light of recent debates about definitions of pre-RA,20–22 the question arose whether the term ‘diagnosis of RA’ is limited to patients with the full picture of the disease or also includes ‘suspected’ RA. However, the majority of the participants felt that ‘suspected RA’ is a field of research with too many uncertainties; that the whole evidence base of RA-treatment rests on a diagnosis (observational studies) or classification of RA (randomised controlled trials, RCTs); and that therefore the management of RA should pertain to those in whom a compelling clinical diagnosis of RA (not necessarily the full classical picture of RA, that we nowadays rarely see) has been made. In this light, the question was also beyond the assignment of this task force but rather appropriate to be dealt with by the task force on the management of early arthritis, whose last update was developed in 2016.23 Consequently, no change was made to this item, it received 100% of the votes and the LoA was 9.9±0.2.
Treatment should be aimed at reaching a target of sustained remission or low disease activity in every patient. While there was general agreement with this recommendation, certain questions arose. One of them related to the positioning of remission before low disease activity in the text, given that most patients in practice have established disease and in those low disease activity would be the prime target. However, the EULAR recommendations attempt to follow a logical sequence, which has been to first address a new patient (phase I of the algorithm in figure 1), then a patient in whom a csDMARD has failed and finally a patient in whom a bDMARD or tsDMARD has failed. Hence, since remission is the main target for patients with early disease, remission was placed before low disease activity. ‘Sustained’ remission or low disease activity refers to the maintenance of this state for at least 6 months. While particularly relating to established disease, in some cases of early RA low disease activity may be also be an acceptable therapeutic target.
A point worthy of mentioning in this context is the definition of remission. Since ACR and EULAR provided Boolean-based and index-based remission criteria already a dozen years ago,3 these criteria have been implicitly integrated in all EULAR Task Forces, as pertinent. Importantly, though, and in line with the above and previous notions on the potential limitations of the patient global assessment (PGA) in the context of defining remission,24 after the meeting it became known that ACR and EULAR have endorsed an increase of the PGA threshold in the Boolean definition of remission from 1 to 2 cm on a 10 cm VAS, while continuing to keep swollen and tender joints at a maximum of 1 and C reactive protein (CRP) at a maximum of 1 mg/dL,25 allowing more patients to be defined as in remission without jeopardising good radiographic and functional outcomes,26 a requirement mandated when that task force was set in place.3
Another question raised by the patient research partner during the deliberations related to the issue of tender joint counts (TJC) and PGA in the context of fibromyalgia accompanying RA. As patients may not find it easy to distinguish which of their symptoms are caused by their RA and which by chronic widespread pain, it was mentioned that these scores may be higher, thus preventing patients reaching the defined state of remission when using instruments scoring disease activity that include the TJC and/or PGA. Others argued that item 5 of the updated treat-to-target recommendations very clearly states: ‘The choice of the (composite) measure of disease activity and the target value should be influenced by comorbidities, patient factors and drug-related risks’ and fibromyalgia is explicitly mentioned in this context.4 27 Thus, one simply needs to adhere to the pertinent recommendations to resolve this question. Moreover, not only the PGA but rather every component of available disease activity instruments may be subject to inconsistencies under certain circumstances: the PGA may be influenced by concomitant fibromyalgia and other pain syndromes (chronic widespread pain); swollen and TJCs may be influenced by the concomitant presence of (inflammatory) osteoarthritis; and acute phase reactants (APRs) like CRP and other biomarkers comprising APRs may respond independently of clinical improvement when antibodies to the IL-6 receptors, JAK inhibitors and even TNF-inhibitors are used28–30; and can of course also be elevated by drivers of inflammation that are independent of RA activity, such as infections. Therefore, attention should be paid to every single item in addition to the global score before adapting therapy.
While overtreatment or better: mistreatment due to misdiagnosis should always be considered,31 it is difficult to quantify what the true frequency of overtreatment is, since no reliable data exist in this respect, while undertreatment was recently shown to be a major problem in RA.32 Importantly, low disease activity rather than remission is the prime therapeutic target in patients with established RA.27 Consequently, some Task Force members felt that even for an established patient with RA with fibromyalgia the current landscape of recommendations leaves little space to misjudge a disease activity state or apply instruments inappropriately, if these recommendations are adhered to, allowing the best outcomes for individual patients to be reached. It was also assumed by some that rheumatologists are capable of differentiating between RA activity and fibromyalgia or other potentially confounding matters, an aspect that lends further support to the importance of overarching principle C.
This item was agreed on by 97.8% of the votes with 1 abstention. LoA was 9.8±0.4.
Monitoring should be frequent in active disease (every 1–3 months); if there is no improvement by at most 3 months after the start of treatment or the target has not been reached by 6 months, therapy should be adjusted. Only little discussion arose when this recommendation was addressed. However, it was specifically demanded that the previous task force’s recommendations to use only instruments that include swollen joint counts for follow-up assessment of disease activity should be reiterated. It was also noted that in many countries swollen (and tender) joint counts are assessed by various well-trained, experienced health professionals rather than rheumatologists. All task force members agreed to keep this recommendation unchanged and it received an LoA of 9.5±0.7.
MTX should be part of the first treatment strategy. In the context of this and the subsequent recommendation (to prescribe leflunomide or sulfasalazine when MTX is contraindicated) the question regarding the application of hydroxychloroquine arose, just as during previous task forces’ deliberations. However, reference was made to an RCT published more than 30 years ago, which very clearly showed a substantial difference in progression of joint damage between sulfasalazine and hydroxychloroquine,33 suggesting that the latter may only be a very weak DMARD. Thus, hydroxychloroquine may be used in patients with early, mild disease (ie, without poor prognostic factors) in whom the other three csDMARDs are contraindicated or not tolerated. Of note, hydroxychloroquine is widely used in other diseases, especially SLE,34 but not for the purpose of inhibiting joint damage progression. Consequently, this drug is not mentioned among the recommendations, because the task force did not wish to suggest that MTX could be replaced by hydroxychloroquine.
Hydroxychloroquine is also frequently used when csDMARD combinations are applied, such as triple therapy with MTX plus sulfasalazine and hydroxychloroquine. This strategy has been shown in some previous studies and SLRs to not provide any added benefit but rather convey more adverse events, leading to low persistence rates.35 Since some rheumatologists continue to use triple therapy as an initial treatment modality, the term ‘part of’ was kept in the recommendation, even though the preference of the current and previous Task Forces is on MTX monotherapy in combination with short-term GCs (see below); however, MTX should be used in any case, unless not tolerated or contraindicated, such as in patients with significant renal impairment.
The Task Force had also no route-of-administration preference, although costs have to be considered in line with overarching principle E. Regarding dose and escalation of csDMARDs, it is suggested to refer to previous recommendations where this was addressed in detail. In brief, in the presence of sufficient folic acid supplementation, MTX can be rapidly escalated to about 25 mg once weekly (in line with a relative dose of 0.3 mg/kg body weight for a person of about 80 kg; lower weekly doses in Asia); sulfasalazine has been previously recommended at a dose of 3000 mg per day and leflunomide at a dose of 20 mg per day without loading dose.
The voting led to 100% agreement with the recommendation, the LoA amounted to 9.6±0.8.tas
In patients with a contraindication to MTX (or early intolerance), leflunomide or sulfasalazine should be considered as part of the (first) treatment strategy. This item continued to receive a high LoA, although the question was raised if the term ‘early’ was needed when speaking of intolerance, since any intolerance constitutes a reason for change. However, since this term had been added many years ago with the implication that "early intolerane" would preclude a judgement on the efficacy of MTX and under these circumstances the replacing csDMARD would still be regarded as a first treatment, with a focus on early disease, and as a counterpart to the term ‘contraindication’, it was decided to leave this point as it was. Again, 100% of the participants agreed with this recommendation, which achieved an LoA of 9.1±1.2.
Short-term GCs should be considered when initiating or changing csDMARDs, in different dose regimens and routes of administration, but should be tapered and discontinued as rapidly as clinically feasible. This is the first recommendation that was changed based on a lengthy discussion. Compared with 2019, the words ‘and discontinued’ were added. Over the last years, the use of GCs was increasingly recommended until it became a strong recommendation to use MTX plus (‘+’) GCs. However, ‘short term’ was always an additional mandate and, it must be reiterated that chronic use of GCs is neither meant nor suggested by this recommendation; therefore, ‘short term’ has been placed at the beginning of this item and the time frame is cleary defined as not more than 3 months (table 1). While in 2019 and in previous versions of this document the term ‘tapering’ always had the meaning of a reduction of the GC dose to zero, semantically, tapering is often interpreted as dose reduction, meaning decreasing rather than stopping medication.
The SLR on the use of GCs has clarified that in all studies in which a reduction and stopping scheme was mandated (prespecified), 90% of patients had indeed stopped GCs, and only about 10% were still on GCs after 24 months.14 A meanwhile partly published, individual patient data meta-analysis reporting about 10% of GC use at 6 to 12 months after the end of the bridging scheme was also presented to the Task Force.36 However, these data come from clinical trials, while in real life, as seen in most registries, chronic GC therapy is used in about half of the patients37–39 and, therefore, the updated recommendations call more strongly than ever before to discontinue GCs as rapidly as possible. Inability to discontinue GCs due to persistently active disease suggests that the ongoing DMARD therapy is not sufficiently effective and needs to be amended, in line with the treat-to-target approach that is also strongly recommended by the EULAR Task Force. The SLR on efficacy16 confirmed the excellent efficacy of a combination of csDMARDs with GC as for instance evidenced in the NORD-STAR trial: non-inferiority was shown for csDMARDs+GC versus certolizumab+MTX and tocilizumab+MTX while abatacept+MTX was statistically superior,40 but in this respect it is important to refer to two other trials, namely AMPLE, comparing abatacept with adalimumab41 and EXXELERATE, comparing certolizumab pegol with adalimumab,42 with superimposable results in both studies. Thus, also NORD-STAR revealed clinical similarity between csDMARD+GC therapy and any bDMARD+MTX treatment, with high rates of stringent remission by CDAI at 24 weeks (>40%) for all these therapies.40 Thus, also notions that ACR-EULAR remission can be achieved only rarely are refuted by NORD-STAR. Overall, the Task Force felt strongly, that this recommendation should be upheld but that the discontinuation of GC should be more strictly advised, as is now done. Consequently, rheumatologists are urged to either apply a single parenteral dose of GCs, such as parenteral (intramuscular) methylprednisolone, as a bridging therapy or predefine a tapering and discontinuation scheme when starting oral GC, with stopping GC to be planned upfront within 3 months; by that time, the csDMARD, such as MTX, should have already shown its efficacy. If patients still require GCs on top of csDMARDs to control disease activity, then the ongoing treatment approach should be considered as insufficient and therapy should be changed. If bDMARD therapy is then indicated, GCs should be discontinued, since the combination of bDMARDs plus GC not only unnecessarily extends the duration of GC therapy, but also is associated with more adverse events, such as infections—any dependence on GCs for more than 4 months should be regarded as definitive failure of the respective DMARD. With so many therapies currently available, it should be feasible, at least in affluent countries, to find the right DMARD-treatment, ultimately allowing all patients to stop GCs. On the other hand, the recent GLORIA trial suggests that low-dose, GC therapy over 2 years may not only be efficacious, but also safe in elderly patients, although long-term data are still missing43; also, it must be borne in mind that major safety concerns of GCs (CV diseases, infections, fractures) occur after more than 5 years of use and, therefore, further data from this trial must be awaited. Overall, the place of GCs is not yet resolved in all its facets. Generally, the Task Force felt the term ‘short-term GC treatment’ would apply to GC use for up to 3 months, while ‘long term’ should refer to a treatment duration of 4–6 months. Any use of GC for more than 6 months should be considered ‘chronic GC treatment’ and therefore be designated as such. Importantly, the Task Force recommends GC-bridging when initiating or changing ‘csDMARDs’, which clearly dismisses the use of GCs when bDMARDs or tsDMARDs are used; indeed, bDMARDs and csDMARDs help to avoid chronic GC use and GCs should be discontinued rapidly after their initiation. Thus, also when csDMARDs are changed or initiated in the presence of bDMARDs or tsDMARDs, use of GCs is not warranted.
The Task Force also found that the SLR had not revealed new safety concerns and that the risks of GC, including CV risk, are well established. That up to 60% of patients in registries44–46 and also patients entering RCTs of new drugs in patients with early or established RA are already on GCs as maintenance therapy may be explained partly by either continuing an insufficiently effective DMARD or by lack of adherence to the recommendation on GC cessation by both patients and rheumatologists. Thus, while the Task Force does not recommend adding GCs when starting a bDMARD or tsDMARD (left part of phase II in the algorithm in figure 1), it does recommend GCs when starting another csDMARD (right part of phase II in figure 1).
Another issue relates to flare therapy. The Task Force is of the opinion that GCs are appropriate flare medications, especially if injected locally into a joint. On the other hand, a flare usually suggests that the DMARD is not sufficiently controlling the disease. Thus, if it is a monoarticular or oligoarticular flare, local GC application may be sufficient for control, but if it is a persistent, polyarticular flare, the DMARD therapy should be reassessed. In particular, GCs should not be instituted instead of an escalation to targeted therapies. In this respect, please see also subsequent comments regarding GC use in low income countries.
With respect to the safety of GCs, two important question could not be answered and become part of the research agenda: when studies refer to cumulative doses, does the duration of GC treatment matter? In other words: is the risk the same if patients receive 1200 mg of prednisone (or equivalent) over 3–4 months (ie, short to long-term use) when compared with the same total dose applied intermittently or over 5 years (chronic use)? There were also calls for better education; doctors, patients, health professionals, should better understand the rationale for using GCs as a bridging strategy and realise how important it is to discontinue the GCs. Research should identify barriers and facilitators for discontinuation of GCs. How can we make it more feasible to taper and stop GCs rapidly to reduce long-term use?
The second issue raised relates to a potential bias by indication in registry patients: do rheumatologists treat patients with certain comorbidities preferentially with GCs chronically because they preclude advancement of targeted therapies? Are such comorbidities possibly related to GC safety issues?
A final point of debate involved patients who had been using GC chronically for years—and how to manage this situation? Indeed, as mentioned above, up to 60% of patients with RA in real life use GCs chronically. Further, some patients may self-medicate high doses in case of perceived disease flare and abruptly reduce the dose when improved. Such an approach may lead to further flaring and may jeopardise the success rate of additional treatment options. In these patients, a slow tapering and cessation regimen may have to be applied individually, and another DMARD should be prescribed on flare. Ideally, patients should not be dependent on GCs to control disease activity in this decade where there are more than a dozen effective DMARDs available. However, this concept is not established and patients who may be dependent on chronic GC use have not been sufficiently studied, another important aspect for the research agenda. Of note, EULAR has developed points to consider for managing difficult-to-treat RA47 and patients who need chronic low doses GCs are obviously ‘difficult to treat’. The Task Force is also aware that in countries with poor resources and thus little or no access to targeted therapies, the chronic use of GCs may be the only way to control patients’ disease activity and quality of life. More studies are needed in people with RA from these low income countries, although the availability of biosimilars and generic versions of tsDMARDs may hopefully alleviate this problem.
The strong reiteration of this recommendation in its amended form by the Task Force is reflected by the 91.3% of ‘yes’ votes with 8.7% abstentions and no vote against it. Also, the LoA of 9.3±1.2 was the highest ever given to a recommendation regarding GCs.
If the treatment target is not achieved with the first csDMARD strategy, in the absence of poor prognostic factors, other csDMARDs should be considered. This recommendation remains unchanged and continues to be mostly based on expert opinion. Clearly, there is a need to perform prospective studies and this point has again been placed into the research agenda. Thus, it appears to be a local political problem if after insufficient efficacy of MTX another csDMARD ought to be be used at all, and this should be discussed between the rheumatological societies and the payers. While the EULAR recommendations must be data driven and widely applicable and cannot account for political issues in individual countries, sometimes - such as here - compromises have to be made. Importantly, the EUAR recommendations can be applied as a template for national recommendations and also used to address controversial views of administrators.
Another discussion point relates to combinations of csDMARDs, such as ‘triple therapy’. Based on available evidence, it was decided many years ago that the EULAR recommendations would not advocate these combinations for reasons already stated above, but they also do not strongly recommend against such strategies. These recommendations are meant as a guidance document prepared by numerous experts in the field, but in daily practice and in front of an individual patient the individual rheumatologist must arrive at the best decision together with the patient.
The recommendation as worded in 2019 and before was approved by 97.8% of the voters with 1 abstention; the LoA was only 8.6±1.4, which is the lowest among all recommendations, likely reflecting the lack of sufficient evidence for it.
If the treatment target is not achieved with the first csDMARD strategy, when poor prognostic factors are present, a bDMARD should be added; JAK-inhibitors may be also considered, but pertinent risk factors* must be taken into account. In 2019, JAK inhibitors were considered at a similar level as bDMARDs in terms of effectiveness and safety. However, based on the data of the ORAL-Surveillance trial among patients with RA>50 years of age with cardiovascular risk factors,7 in which more major adverse cardiovascular events (MACEs) and higher malignancy rates with tofacitinib compared with TNF-inhibitors were observed, a change of this recommendation was required. While similar findings were not reported from long-term extension trials and registries,48 49 results of a single RCT convey a higher LoE compared with other non-trial data; moreover, the trial was performed in a population with specific risk factors.
The Task Force arrived at the above formulation after discussion of many pros and cons regarding the use of JAKi. These deliberations are detailed below so that readers can follow the way to the decision. In brief, the majority of the Task Force members were of the opinion that the data on risks due to tofacitinib currently pertain only to patients at risk and that these risk factors should be clearly communicated. On the other hand, the Task Force found no evidence for greater risk of tofacitinib versus TNFi in patients without risk factors. While data for other JAKi do not exist beyond registers and long-term extensions of clinical trials, one cannot exclude that a similar risk could also be associated with non-tofacitinib JAKi when subjected to an outcomes RCT. The previous recommendation was amended based on these considerations.
As always, the EULAR Task Force wishes to be transparent with respect to the process that led to its decision and presents details of the discussions about this recommendation. These were lengthy, because many questions were raised, most of which could not be answered by the SLR data or by the experts present in the room. These questions included: (1) Should a new recommendation interpret the ORAL-Surveillance data as relevant only for tofacitinib, or—in the absence of exonerating RCT data for the other JAKi—as relevant for the entire class of JAKi? (2) Should JAKi be fully eliminated from treatment-phase II and only be recommended for use after bDMARDs have failed? The US FDA has decided along this line50 and suggested to use JAKi only after TNF-inhibitors have failed. (3) Given the abundance of available bDMARDs, should we reserve JAKi for use only after all bDMARD modes of action have failed, in other words: should we create a phase IV in the treatment algorithm?
All these points were addressed in detail and several proposed amendments of the recommendations were discussed. In this respect, also the patient research partner’s views were of particular importance. These comments related to the importance of shared decision making especially under the circumstances of the ORAL Surveillance data, and the advantage of having more therapeutic options available with different modes of action and routes of administration as long as the perceived benefits outweigh the perceived risks. Transparency is key here; a patient can only make an informed decision after being fully told about the benefits and risks. Of note, the patient research partner specifically addressed the importance of not deterring future new drug development as a consequence of restricting the use of all JAKi based on one study solely related to tofacitinib and that it was important JAKi are still prescribed in order to accumulate real-world data on their safety.
When it came to voting on a new or amended recommendation, several options were discussed. The proposal to change ‘tsDMARDs’ into ‘JAK1/2 inhibitors’ or only ‘JAK1 inhibitors’ received so much opposition that it was not further pursued for voting; most of the Task Force members thought that it was currently not possible to make statements on higher or lower risks of JAKi based on their (theoretical) selectivity.
The first voting round then took place between two options. Option 1 proposed to delete ‘or a tsDMARD’ in recommendation 8 and leave this only for use after a bDMARD has failed, in other words to move JAKi to phase III of the treatment algorithm; option 2 suggested placing a semicolon after the current recommendation and then adding: ‘but bDMARDs should be favoured over JAKi in those with pertinent risk factors’. Option 1 received 32% and option 2 attained 68% of the votes. While this voting-result revealed a clear preference, the majority needed for this first round (75%) was not met.
In the subsequent discussion, it was proposed to develop a dual message in this recommendation, to delete ‘or a tsDMARD’ from it and separate the remains from the subsequent statement by a semicolon. The subsequent part would then either read: ‘JAK-inhibitors may also be considered in patients without pertinent risk factors*’ (option 3); or: ‘JAK inhibitors may also be considered in the appropriate patient taking pertinent risk factors* into account” (option 4), with the asterisks defining risk factors in a footnote. Option 3 received 23% of the votes, and option 4 received 72% of the votes, while 5% abstained. Thus, option 4 was agreed to by the appropriate majority, and after some wordsmithing it was formed into the new recommendation 8 as stated above and in table 2. Recommendation 8 now clearly separates bDMARDs from JAKi, but still does not fully refute JAKi at this stage of the treatment cascade. Rather, it calls for a considerate approach to the use of JAKi and mandates a careful evaluation of the risks that individual patients may carry with them, as well as shared decision making after fully informing the patient. The term ‘may be considered’ in conjunction with the ‘must’ regarding assessment of pertinent risk factors best reflects the thinking-process of the Task Force regarding this recommendation.
Finally, a discussion on the risk factors ensued. Was 65 years the appropriate age? What about patients who started a JAKi at age 62 and reached 65 during the course of treatment—would they then have to stop therapy? Are smokers who stopped 10 or 20 years ago at the same risk for malignancy as current smokers? After some discussion on the definition of risk factors, it was decided to focus primarily on the risk definitions mentioned by the European Medicines Agency (EMA), these were added as a footnote to recommendation 8 and comprise age over 65 years, history of current or past smoking, other cardiovascular risk factors, other risk factors for malignancy, and risk factors for thromboembolic events (details are mentioned in the respective footnotes to table 2 and figure 1).51
Of note, the focus on CV and malignancy risk here is a consequence of the recently published data, but it is evident since the introduction of bDMARDs more than 20 years ago that infection risks, especially risks of tuberculosis reactivation or Herpes zoster, has to be taken into account and respective precautious measures initiated as needed. Moreover, in a substudy of ORAL-Surveillance, published several months after the Task Force meeting, an increased infection rate beyond Herpes Zoster was seen for tofacitinib compared with TNF-inhibition.52
Regarding dosing of b/ts DMARDs, the Task Force refers to previous versions of this manuscript and various consensus statements, such as starting with 8 mg/kg of tocilizumab rather than 4 mg/kg, if intravenous dosing is preferred, or the use of 2×500 mg or 1×1000 mg rituximab rather than 2×1000 mg. Further, in the absence of contra-indications (see above), for baricitinib, the 4 mg daily dose, as approved in Europe, has some efficacy advantages especially in patients with long-standing RA compared with the 2 mg daily dose as approved in the USA. Finally, the use of loading doses for certolizumab pegol or sc abatacept may have to be revisited.
The recommendation achieved 100% approval and this is a good example of how discussions and exchanges of thought can lead to a compromise that is viable for everyone in spite of initially opposing views. Consequently, the subsequent LoA of 9.1±1.1 was high.
Many questions raised during the deliberations were considered important topics for the research agenda (box 1).
bDMARDs and tsDMARDs* should be combined with a csDMARD; in patients who cannot use csDMARDs as comedication, IL-6 pathway inhibitors and tsDMARDs* may have some advantages compared with other bDMARDs. No new compelling evidence was gained regarding monotherapy of bDMARDs or tsDMARDs compared with combination therapy. Therefore, the EULAR Task Force continues to advocate the continuation of MTX (or other csDMARDs) when treatment with bDMARDs or JAKi is planned. In this context, it should be borne in mind that once patients have arrived at this stage, they usually have tolerated MTX well and do not need to stop the drug due to intolerance. Moreover, as also repeatedly stated in previous versions of the recommendations, the MTX dose can be reduced to as low as 10 mg weekly to convey the added benefit of combination vs monotherapy.53 54
No textual change was made in this recommendation, which received 100% of the votes and attained an LoA of 9.2±0.9; however, asterisks were added after the words ‘tsDMARDs’ to account for the risk factors addressed in recommendation 8, as will also be done in subsequent recommendations as pertinent.
If a bDMARD or tsDMARD* has failed, treatment with another bDMARD or a tsDMARD* should be considered; if one TNF- or IL-6 receptor inhibitor therapy has failed, patients may receive an agent with another mode of action or a second TNF- or IL-6 receptor inhibitor. Since the SLR revealed that sarilumab can replace tocilizumab and is efficacious also in patients in whom tocilizumab has failed, thus partly answering a previous research question, the old recommendation could be expanded to include IL-6R inhibitors rather than just mentioning TNF-blockers, although only observational or extension data exist for IL-6R inhibitors,55 56 while RCTs have been performed with TNF-blockers.57 58 On the other hand, we still miss data on the efficacy and safety of using a JAKi after another JAKi has failed and this, again, is part of the research agenda. Also, patients who have failed multiple b/tsDMARDS have to be seen as difficult-to-treat RA in line with the respective EULAR definition and points to consider.47 59 Almost 98% of the participants voted for this change with no one against it. The LoA was 9.3±0.8.
After GCs have been discontinued and a patient is in sustained remission, dose reduction of DMARDs (bDMARDs/tsDMARDs and/or csDMARDs) may be considered. This new recommendation has been constructed by combining the last two items from 2019 which read as follows: ‘If a patient is in persistent remission after having tapered GCs, one can consider tapering bDMARDs or tsDMARD, especially if this treatment is combined with a csDMARD.’ And: ‘If a patient is in persistent remission, tapering the csDMARD could be considered.’5 Evidence has emerged indicating that there was no difference in clinical outcome when either a bDMARD or csDMARD was tapered first. It had previously been suggested to start with a reduction of bDMARDs because of the costs involved. However, an economic analysis has revealed that the total costs of tapering csDMARDs first vs tapering anti-TNFs first did not differ.60 Consequently, the Task Force was of the opinion that there is no preferred tapering sequence and this can be left to the discretion of patients and rheumatologists in a shared decision, but still with an open eye on costs, since prices of bDMARDs may vary significantly within and between countries.
In addition, the place of GC tapering was changed. As discussed above for item 6, the term tapering is often misinterpreted and, therefore, the Task Force stipulated that GCs must be ‘discontinued’ before considering tapering other agents. For that reason, the GC part of this recommendation was moved to the beginning of the recommendation.
Importantly, though, there is also compelling evidence that stopping bDMARDs and/or csDMARDs will ultimately lead to flares in most patients.61–63 Therefore, the Task force felt that either dose reduction or interval increase (‘spacing’) is preferred, but completely stopping may not be advisable. Of note, most (though not all) patients who flare after dose reduction can be brought back into a good disease state after reintroduction of the original dose. Also, as discussed in previous versions, tapering of DMARDs should only be started if a patient is in persistent stringent (ACR-EULAR) remission for at least 6 months, although more data may be needed to determine the lowest level of disease activity that provides a good prediction for maintenance of a good state. Finally, it was noted that tapering trials were very heterogeneous and that some standardisation by regulators or professional societies would be needed.
This new recommendation received 95.4% of the votes; 2.3% abstained and 2.3% voted against. The LoA amounted to 9.3±1.1.

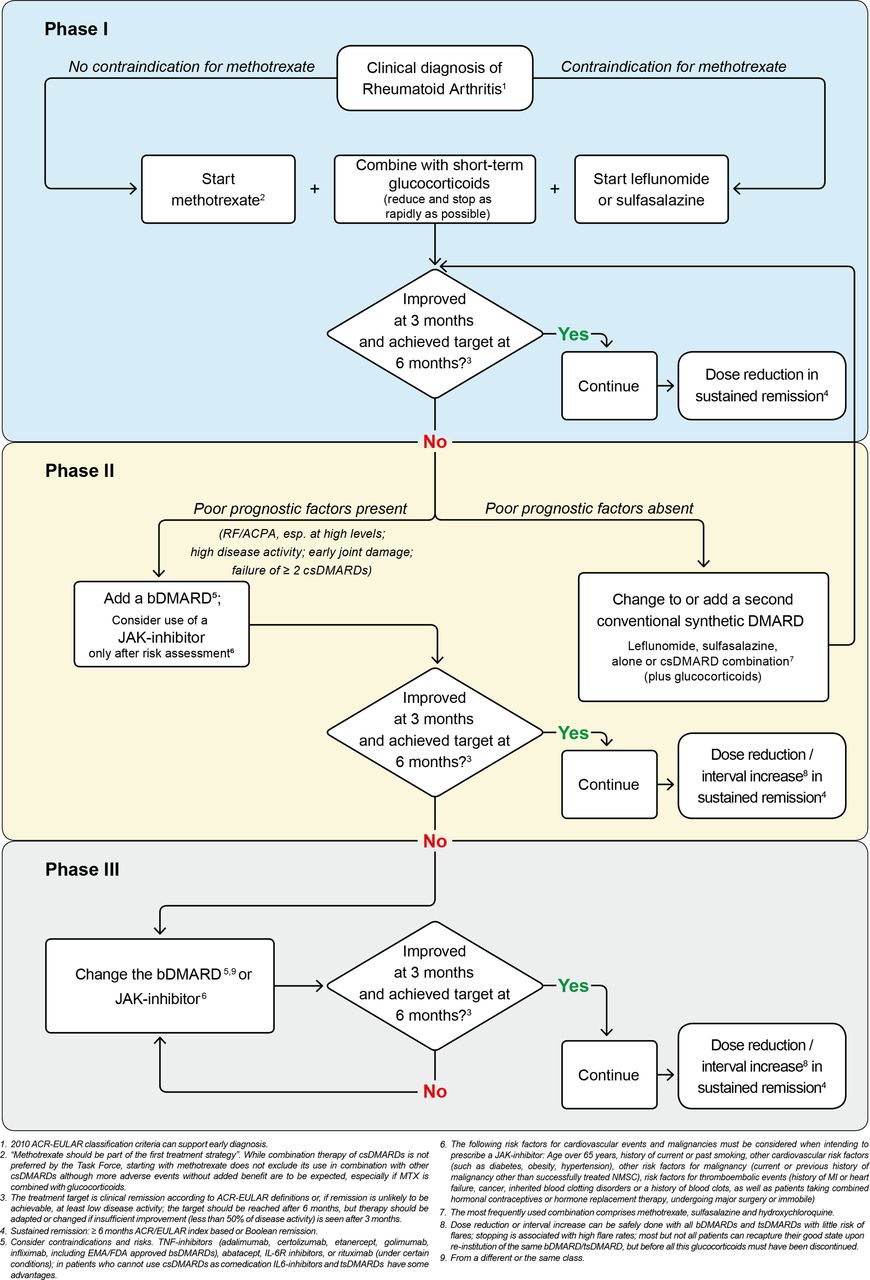{kind=link}
Flow chart. ACR, American College of Rheumatology; bDMARDs, biological disease-modifying antirheumatic drugs; csDMARDs, conventional synthetic DMARDs; FDA, Food and Drug Administration; JAK, Janus kinase; MTX, methotrexate;NMSC, non-melanoma skin caner; tsDMARDs, targeted synthetic DMARDs.
Research agenda
Glucocorticoids
Is the risk of glucocorticoids (GCs) different if a specific cumulative dose has been used within a relatively short period of time, such as up to 3 or 6 months, or chronically over a number of years?
What are the barriers and facilitators of GC cessation after induction therapy and how can a strategy for tapering and discontinuing be best implemented?
Does the concomitant use of GCs at very low doses (1–3 mg prednisone equivalent) increase therapeutic success without producing unacceptable side effects?
Can the chronic use of GCs be prevented by rapid (ie, within 3-6 months) switching of disease-modifying antirheumatic drug (DMARDs) in patients who have active disease despite DMARDs of whatever kind?
How frequent is the chronic use of GCs among patients with rheumatoid arthritis (RA) followed in resource poor countries and how could such chronic use be mitigated or prevented?
What are the effectiveness and safety profiles of (repeated) intramuscular glucocorticoids, for example, methylprednisolone 120 mg or triamcinolone 80 mg 1–4 times yearly?
Are safety issues with chronic GC use related to pre-existing comorbidities and do patients with such comorbidities preferentially receive GCs rather than advancing to biological/targeted synthetic (b/ts) DMARD therapies?
Janus kinase (JAK)-inhibitors and bDMARDs
To which extent do in vitro selectivity and in vivo selectivity differ among JAK inhibitors (JAKi)?
Are the cardiovascular and malignancy risks of JAKi as seen in the ORAL-Surveillance study, different with JAK-1 or JAK-1/2-selective agents than with pan-JAKi?
Which mechanisms lead to the cardiovascular events and the increase in malignancies seen with tofacitinib?
Which mechanisms lead to the increased risk of thromboembolic events with JAKi?
Is monotherapy of JAKi or combination of JAKi plus methotrexate (MTX) more efficacious than MTX+GC? Ideally, an active control arm using a TNF-inhibitor (TNFi) or tocilizumab (plus MTX) should be included in such a trial
How safe and efficacious is the use of a JAKi after another JAKi has failed?
How safe and efficacious is the combination of a JAKi with a bDMARD, such as a TNFi, in patients who have failed to respond to multiple drugs?
How safe and efficacious is the use of an IL-6 pathway inhibitor if a JAKi has failed?
How safe and efficacious are abatacept, tocilizumab and rituximab after any of the other non-TNFi bDMARDs or a tsDMARD has failed?
Treatment strategy
Can we identify new biomarkers to stratify patients and to predict therapeutic response or lack of response?
Is tapering of bDMARD monotherapy possible?
Will randomised controlled trials on tapering of bDMARDs and tsDMARDs, designed to following predefined predictors for maintenance of good outcomes after their withdrawal, show success?
How good is patient adherence to a bDMARD or tsDMARD and can non-adherence explain secondary loss of efficacy?
How long should the duration of persistent remission be before conventional synthetic (cs)DMARDs can be tapered?
Can taxonomy of RA be improved to guide therapeutic decisions?
Can the identification of disease phenotypes inform tailored therapeutic use?
Will therapeutic drug monitoring improve disease course and outcome and support decisions about switching within or between drugs?
Is leflunomide equivalent to MTX as first line csDMARD therapy?
Is there true secondary loss of efficacy or is this due to non-adherence? And if the former, what is the reason for this loss of efficacy?
What is the optimal treatment target: remission or low disease activity?
What is the true frequency of undertreatment and that of overtreatment in RA clinical settings?
Risk stratification for DMARD use
Does the risk stratification for bDMARD/tsDMARD initiation based on presence of good or bad prognostic factors as recommended by EULAR translate into improved outcomes for both prognosis groups?
Do patients who lack poor prognostic factors benefit as much from a switch to or addition of a csDMARD as from the addition of a bDMARD?
Difficult to treat RA
What is the optimal treatment approach to refractory RA?
Which other factors (eg, life-style characteristics, treatment history) allow the best possible therapeutic decisions to be made?
Pre-RA
What is the optimal (therapeutic) approach to arthralgia suspicious for progression to RA?
All overarching principles and recommendations are summarised in table 2 together with respective footnotes for specific definitions, LoEs, grades of recommendation and LoA. An abbreviated, graphical form of the recommendations is presented in figure 1, also together with respective footnotes. The explanatory part for each individual recommendation in the manuscript is part of the recommendations which only stand in full when these explanations and expansions are taken into account.
A research agenda is shown in box 1. Some points of the old questions have been worked up, others wait to be addressed soon and are repeated here. As indicated above, many new areas for research were opened during the meeting and this is also reflected in box 1.
Discussion
In contrast to previous years, new drug classes have not emerged since the last update of the recommendations. However, the Task Force focused on the new data, particularly safety data for existing drugs, a process that led to significant changes of the 2019 recommendations. At the same time, all overarching principles as well as 6 of the 11 recommendations remained unchanged, which testifies to the validity and maturity of these previous recommendations. However, the LoE for recommendation 7 remained low and to improve its LoE, well-designed trials with the main outcome focusing on the question of the optimal therapy for patients with a low risk of joint damage progression and insufficient response to MTX+GCs are needed.
In line with previous versions of this document, the recommendations adhere to a logical sequence, which starts with a focus on newly diagnosed patients and provides guidance along the disease course and treatment history of the patients. Of course, if a patient has already established disease and these recommendations are consulted, then the treatment path will start at the pertinent point, such as phase II for insufficient responders to MTX and/or another csDMARD or phase III for insufficient responders to a bDMARD or JAKi. Thus, these recommendations can be applied for any patient at any point in time.
It is noteworthy that this year’s set of recommendations is the smallest ever. While in 2010 fifteen recommendations were compiled,1 these were reduced in a stepwise manner to 12 items in 2019 and to 11 in 2022. These reductions are not deliberate or primarily driven by parsimony; rather, they are a logical consequence of accumulating evidence. Of note, the accumulating evidence encompassed significant parts of research agendas, presented in previous versions. Increasing evidence enables a greater focus on what is important, possibly yet another consequence and advantage of the strategy taken to use the Oxford Evidence-Based Medicine approach rather than others. The clearer the information provided in recommendations, the better and easier they may be followed by clinicians.
Three small and one major changes to the recommendations were implemented. The first small change relates to the use of GCs as bridging therapy, when a csDMARD like MTX is started. While already previous Task Forces clearly recommended only short-term use of GCs with rapid tapering and cessation, this may not have been phrased clearly enough. Therefore, recommendation 6 now explicitly and unequivocally advocates not only a rapid tapering regimen but also timely discontinuation. To this end, physicians should make clear to patients at the time of first prescription that GCs are only a bridging therapy and physicians and patients should liaise to adhere to a prespecified discontinuation strategy. There may be some reasoning behind a preferential use of parenteral GCs in this respect, as physicians can control their timing and dosage. The efficacy of GCs as an adjunct to csDMARDs continues to be unsurpassed as revealed in the NORD-STAR trial: no bDMARD plus MTX shows a major clinical benefit over GCs plus MTX.40 Consequently, the current Task Force adhered to the general principles of this recommendation that contrasts with the most recent guideline of the ACR which recommend not using GC even as a bridging therapy.8 It is noteworthy that JAKi have not yet been assessed against MTX plus GC; this is another point for the research agenda.
The second small change accounts for the fact that IL-6R inhibition has now been tested after insufficient response to another IL-6R blocker.55 56 This led to including IL-6R blockade in addition to TNF-inhibition in patients in whom a previous bDMARD with the same mechanism of action has failed in recommendation 10. A study published after the meeting supports the Task Force’s view that a JAKi may be efficacious after another JAKi failed, although this observation is limited to registry data.64 This is to be addressed in more detail in future research activities.
The third small change occurred when previous recommendations 11 and 12 were brought together and relates to the topic of tapering drugs in patients with sustained remission. Of note, when speaking of sustained remission, we refer to previously presented data which suggested not starting tapering before achieving 6 months of stringent remission.5
The most intensive debate and most extensive change occurred for recommendation 8 which previously suggested positioning bDMARDs and tsDMARDs at a similar level, when MTX (plus GC) were not sufficiently efficacious (phase II). The new safety issues emanating from the ORAL-Surveillance trial,7 which answered question 15 of the 2019 research agenda,5 are concerning. While they appear to be at odds with data from registries, they have to be taken seriously since they come from an RCT conducted in a prespecified high-risk population with safety as the primary outcome. While the observed increases in MACEs and malignancies compared with TNF-inhibition were unexpected, the malignancy aspect was particularly concerning, especially given the frequent literature discussions on the risk of malignancies when using TNF-blockers and other bDMARDs65 66 which, however, was not observed in registries.67 68 Of note, in line with the nature of risk factors, increased event rates were seen in patients with high-risk compared with low-risk categories, even when treated with TNF-inhibitors; however, also in the higher risk categories events were still elevated on tofacitinib compared with patients treated with anti-TNFs.69
To date, it is speculative which mechanisms are responsible for the abnormalities seen in the ORAL-Surveillance trial. It is not easy to explain the mechanisms leading to an increase in MACEs, since a recently completed similar RCT, the ENTRACTE trial, showed no increase in MACEs on tocilizumab treatment compared with TNF-blockers.70 This makes the inhibition of IL-6 signalling unlikely to be responsible for the finding in ORAL-Surveillance. Further, tofacitinib is essentially a pan-JAKi; can one extrapolate data resulting from this single agent to other more selective JAKis? Alternatively: can one exclude that more selective JAKis exhibit the same risks? These questions can only be answered by additional outcome studies, and two are indeed currently ongoing, although in patients at risk of thromboembolic events71 72; these data, however, will become available only in the midst of this decade. All these points provide lots of room for further research.
Given the results of ORAL-Surveillance, it was evident that recommendation 8 would have to undergo a major change. No Task Force member felt that the recommendation could stay unchanged. The discussions centred around several scenarios, from excluding JAKis totally from phase II, via separating JAKis from bDMARDs, to modifying the current recommendation so that risks shown in the ORAL-Surveillance trial could be accounted for. The trial's findings related to a patient population of older individuals with certain risk factors for cardiovascular disease being present. In contrast, long-term extension data of clinical trials with tofacitinib49 had excluded patients with relevant risks before the start of the trials, and registry data,48 while including all patients in whom this treatment had started, are likely confounded by indication. An RCT has to be seen as the most important piece of evidence.
On the other hand, the rationale for the warning by the FDA to reserve JAKis only to patients in whom a TNF-blocker had failed was not fully understood by the Task Force: why only after TNF-inhibitors and not after anti-IL-6R antibodies, which have shown to be of no greater risk than TNF-inhibitors, especially since ORAL-Surveillance was not performed in patients who had failed to respond to TNF-inhibitors?70 Why only after one TNF-inhibitor had failed and not after more than one?
Finally, the Task Force arrived at a decision which was unanimously endorsed by its members. Of note, this was one of the largest EULAR Task Forces and spanned the largest array of continents ever, including the representation from Australia and Africa. The endorsed recommendation no. 8 places JAKis at the same level as bDMARDs, but only in patients in whom risk factors for cardiovascular or malignant diseases have been considered specifically, as part of a shared decision making process. This means that bDMARDs, irrespective of their mode of action, should be preferred over JAKi in patients with RA with risk factors for malignancy or MACE. Only in patients without such risk factors, JAKis may be considered instead of bDMARDs. All these risk assessments should be made in agreement with the patient: patients must be informed about the benefits and risks of all drugs and the choice of the treatment should be based on a shared decision, in line with the very first overarching principle.
The Task Force adhered to the three previous phases of the treatment cascade and did not address ‘pre-RA’ or ‘patients at-risk of developing RA’; while ‘pre-RA’ was part of the 2019 research agenda, data on which solid recommendations could be formulated were still not available, and it may be necessary to address this point in an update of the EULAR recommendations for the management of early arthritis.23 On the other side of the spectrum, the Task Force also did not address the management of patients who have failed multiple bDMARDs and/or tsDMARDs, another point of the last research agenda.5 However, meanwhile EULAR has provided a definition of refractory or difficult-to-treat RA59 and, as briefly mentioned above, also points to consider for the management of these patients.47 Importantly, as we lack predictors of treatment response in individual patients, the Task Force currently recommends a treat-to-target strategy that includes cycling between existing b/tsDMARDs in phase III of the algorithm. More data may be needed to develop better evidence-based approaches regarding the recognition and treatment of patients with highly active disease despite many therapies. It has been suggested that this population is increasing in number.73 The next update may then be better able to also address this important aspect.
In summary, the 2022 update of the EULAR recommendations presented here is the fifth version of this EULAR activity and every time a Task Force was convened, new aspects of the management of RA were discussed and respective changes developed—a true rationale for the process of updating recommendations. The current version will inform rheumatologists, health professionals, patients, regulators, payers and other stakeholders on the current views derived during this Task Force’s debates on the presumably best way to treat RA at the beginning of the current decade, a year that also marks EULAR’s 75th anniversary. The RA management recommendations reflect better than many other achievements how far rheumatology has come since the days when EULAR was founded.74 75 And with every new drug, with every new insight and with every update of EULAR’s management recommendations, hopefully more patients will attain the treatment target, ultimately allowing us to state that active disease has been eradicated in RA, just like severe joint damage is hardly seen any more today on adherence to respective treatment strategies. The research agenda presented in box 1 may help to arrive at this state within the next few years—more trials, leading to more insights, and more effective strategies will be needed to get there.
Interestingly, at the end of the discussion section of the 2019 update, we stated that the update had ‘reached a steady state of evidence’.5 In 2022, we learnt that such seemingly steady state can be easily shaken up by new data, teaching us that one needs to continuously track the evolving evidence meticulously, with devotion and without prejudice. Consequently, the evolution of new findings will have to be thoroughly followed, and we suppose that an update of the recommendations may become necessary within the next 3 years.
Ethics statements
Patient consent for publication
References
Supplementary materials
Lay summary
Disclaimer : This is a summary of a scientific article written by a medical professional (“the Original Article”). The Summary is written to assist non medically trained readers to understand general points of the Original Article. It is supplied “as is” without any warranty. You should note that the Original Article (and Summary) may not be fully relevant nor accurate as medical science is constantly changing and errors can occur. It is therefore very important that readers not rely on the content in the Summary and consult their medical professionals for all aspects of their health care and only rely on the Summary if directed to do so by their medical professional. Please view our full Website Terms and Conditions.
Copyright © 2022 BMJ Publishing Group Ltd & European League Against Rheumatism. Medical professionals may print copies for their and their patients and students non commercial use. Other individuals may print a single copy for their personal, non commercial use. For other uses please contact our Rights and Licensing Team.
Footnotes
Handling editor David S Pisetsky
Twitter @AlexSepriano, @Janetbirdope, @cardielmh, @drpnash, @FeliceRivellese
JSS and RBML contributed equally.
Correction notice This article has been corrected since it published Online First. The competing interests statement was erroneously omitted and is now included in the online version only.
Contributors JSS wrote the first draft, RBML continued work and all authors contributed importantly to the manuscript.
Funding This study was funded by European League Against Rheumatism.
Competing interests AB: Grants from Abbvie, BMS, Pfizer, Roche, UCB and honoraria from Abbvie, Galapagos, Lilly, Nordic, Novartis, Pfizer, Sanofi. AAdB: Grants to the institution from Abbvie, Gilead, Galapagos, Lilly, Novartis, Pfizer, Sanofi. FB: Grants from Abbvie, Biogen, BMS, Hexal, Horizon, Medac, Pfizer, Lilly, Sanofi, and honoraria from Abbvie, Biogen, Galapagos, Medac, Pfizer, Roche, Sanofi, Janssen, Lilly. MHB: Institutional grant/research support from Gilead Sciences, Pfizer Inc, and UCB; consultant for AbbVie, Eli Lilly, Galapagos, Pfizer; and member of the speakers’ bureau for AbbVie (with all honoraria paid to host institution; the views expressed are those of the authors and not necessarily those of the NHS, the NIHR, or the Department of Health. CC: Support for clinical trials and scientific projects: AbbVie, Ewopharma, Lilly, Novartis, Pfizer; Speaker fees & consulting: AbbVie, Amgen, Boehringer Ingelheim, Ewopharma, Lilly, Novartis, Pfizer, Sandoz, UCB. KC: Consultancy fees from from Eli Lilly, AbbVie and Pfizer. MC: Speaker, researcher or advisor for Abbvie, Eli Lilly, Gilead, GlaxoSmithKline, Janssen and Pfizer. MC: Research funds and lecture fees from: Alfa Wassermann, Amgen, BMS, Boehringer Ingelheim, Celgene, DS Medica, Horizon, Pfizer. RC: consulting fees from Abbvie, BMS, Amgen, Celltrion, Lilly, Fresenius-Kabi, Galapagos, Janssen, MSD, Novartis, Pfizer, UCB. AF: Honoraria from AbbVie, Astra-Zeneca, Eli-Lilly, Gilead, Pfizer, Viatris; grant support (to the institution) from AbbVie, BMS, Eli-Lilly, Galapagos, Pfizer. JEF: unrestricted research grants or speaker for Abbvie, Biogen, BMS, Janssen, Lilly, Medac, MSD, Novartis, Pfizer, UCB. EAH: Expert advice to and/or speaking engagements for Pfizer, AbbVie, Celgene, Novartis, Janssen, Gilead, Lilly, and UCB. AI: Honoraria, advisory boards, speakers bureau, educational grants, research support from: AbbVie, Alfa-sigma, Amgen, Biogen, BMS, Celgene, Celltrion, Eli-Lilly, Galapagos, Gilead, MSD, Novartis, Pfizer, Sanofi Genzyme, SOBI, UCB. KLH: Honoria from Abbvie and grants from Pfizer and BM. SKL: Honoraria from Pfizer, Celltrion and Viatris. GRB: Honoraria for lectures and consulting: Abbvie, BMS, Chugai, Galapagos, Lilly, Medac, MSD, Pfizer, Sabofi, UCB. IMI: Consulting fees and honoraria from AbbVie, AstraZeneca, BMS, Boehringer Ingelheim, Cabaletta, Causeway Therapeutics, Celgene, Evelo, Gilead, Janssen, Pfizer, Novartis, Lilly, Moonlake and UCB Pharma; Research support from BMS, Boehringer Ingelheim, Celgene, Janssen, Novartis and UCB Pharma. GR: Honoraria from Abbvie, Lilly, MSD, Novartis, Pfizer and Roche. EM: Honoraria as speaker, clinical researcher or advisor from: Astra Zeneca, AbbVie, Pfizer, Lilly, Janssen, Sanofi, Roche, GSK, Amgen, Sandoz, Novartis. PN: Funding for research and clinical trials and honoraria for advice and lectures on behalf of Abbvie, BMS, Pfizer, Lilly, Novartis, Janssen, Gilead/Galapagos, Samsung, GSK, Boeringher Ingelheim, MSD, Celltrion. ARR: honoraria for lectures and consultation from Abbvie, Amgen, BMS, Galapagos, Lilly, MSD, Pfizer, Roche, Sanofi, UCB. HSK: consultant and advisor or review panel member for AbbVie, Amgen, AstraZeneca, Biogen, BMS, Celgene, Gilead, Galapagos, Hexal Sandoz, Janssen-Cilag, Elli Lilly, Medac, MSD, Merck, Novartis, Pfizer, Roche, Sanofi-Aventis, and UCB. IS: Honoraria from Abbvie, Amgen, Pfizer, UCB. AS: Non-personal grants from AbbVie, Amgen, Bristol-Myers Squibb, Celltrion, Galapagos, Hexal AG, Lilly, MSD Sharp & Dome, Pfizer, Roche, Samsung Bioepis, Sanofi-Aventis, UCB, Viatris and speaker honoraria from AbbVie, BMS, Celltrion, Galapagos, Lilly, MSD, Pfizer, Roche. EvD: Honoraria for consultation from Abbvie, Adcock Ingram, Amgen, Boehringer-Ingelheim, Janssen, Lilly, Pfizer, Roche. TT: Grants from AbbVie, Asahikasei, AYUMI, Chugai, Eli Lilly Japan, Mitsubishi-Tanabe, Ono, and honoraria from AbbVie, Asahikasei, Astellas, Bristol-Myers Squibb, Chugai, Daiichi Sankyo, Eisai, Eli Lilly Japan, Gilead Sciences, Janssen K.K., Mitsubishi-Tanabe, Pfizer Japan. TPMVV: Speaker fees from Abbvie. KLW: Consulting for Pfizer, AbbVie, Union Chimique Belge (UCB), Eli Lilly & Company, Galapagos, GlaxoSmithKline (GSK), Roche, Gilead, BMS, Regeneron, Sanofi, AstraZeneca, Novartis; research grants from BMS and Pfizer. RBML: Receiving honoraria for lectures and consulting from AbbVie, BMS, Eli-Lilly, Galapagos, Gilead, Jansen, Novartis, Pfizer and UCB. Owner of Rheumatology Consultancy BV, a company under Dutch law. Providing expert-testimony for AbbVie at EMA. EULAR’s acting chair on Quality of Care.
Provenance and peer review Not commissioned; externally peer reviewed.
Author note After the review process of this manuscript it became known that the Pharmacovigilance Risk Assessment Committee (PRAC) of EMA has provided new recommendations regarding the use of JAKi (https://www.ema.europa.eu/en/documents/referral/janus-kinase-inhibitors-jaki-article-20-referral-ema-recommends-measures-minimise-risk-serious-side_en.pdf). Item 8 of the updated EULAR recommendations presented here is in line with the PRAC recommendations. These recommendations have been presented at the EULAR Congress in June 2022.