Article Text

Abstract
Objectives Rheumatic immune-related adverse events (irAE) such as (poly)arthritis in patients undergoing immune checkpoint inhibitor (ICI) treatment pose a major clinical challenge. ICI therapy improves CD8+ T cell (CD8) function, but CD8 contributes to chronic inflammation in autoimmune arthritis (AA). Thus, we investigated whether immune functional and metabolic changes in CD8 explain the development of musculoskeletal irAE in ICI-treated patients.
Methods Peripheral CD8 obtained from ICI-treated patients with and without arthritis irAEs and from AA patients with and without a history of malignancy were stimulated in media containing 13C-labelled glucose with and without tofacitinib or infliximab. Changes in metabolism, immune-mediator release, expression of effector cell-surface molecules and inhibition of tumour cell growth were quantified.
Results CD8 from patients with irAE showed significantly lower frequency and expression of cell-surface molecule characteristic for activation, effector-functions, homing, exhaustion and apoptosis and reduced release of cytotoxic and proinflammatory immune mediators compared with CD8 from ICI patients who did not develop irAE. This was accompanied by a higher glycolytic rate and ATP production. Gene-expression analysis of pre-ICI-treated CD8 revealed several differentially expressed transcripts in patients who later developed arthritis irAEs. In vitro tofacitinib or infliximab treatment did not significantly change the immune-metabolic profile nor the capacity to release cytolytic mediators that inhibit the growth of the human lung cancer cell line H838.
Conclusions Our study shows that CD8 from ICI-treated patients who develop a musculoskeletal irAE has a distinct immune-effector and metabolic profile from those that remain irAE free. This specific irAE profile overlaps with the one observed in CD8 from AA patients and may prove useful for novel therapeutic strategies to manage ICI-induced irAEs.
- Inflammation
- Biological Therapy
- Arthritis
- T-Lymphocyte subsets
Data availability statement
Data are available upon reasonable request. All data are presented in the manuscript. Raw experimental data will be made available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
Immune checkpoint inhibition (ICI) therapies have a high success rate regarding progression-free and overall survival for patients with cancer. However, up to 20% of ICI-treated patients develop musculoskeletal immune-related adverse events (irAE) that are often associated with severely reduced quality of life.
To avoid precocious ICI treatment termination, strategies to treat rheumatic irAE must be simultaneously efficient in curbing musculoskeletal symptoms without interfering with the antitumor therapy.
CD8+ T cells play a pivotal role both in arthritis pathogenesis and antitumor responses.
WHAT THIS STUDY ADDS
Immunofunctional and metabolic analysis of peripheral CD8+ T cells from patients with musculoskeletal irAEs revealed that they share a common profile with those from patients with chronic autoimmune polyarthritis (AA) but are distinct from ICI-treated patients who remained irAE free.
CD8+ T cells from patients with irAE treated in vitro with the Janus-kinase (JAK) pathway inhibitor tofacitinib and TNF-α blocker infliximab still maintained the capacity to release cytokines and cytolytic molecules, express immune-effector cell surface molecules and prevent the growth of a human lung cancer cell line.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
The specific immunofunctional and metabolic profile in rheumatic irAEs and its overlap to AA-CNT profile is a potential starting point for a better understanding of the pathogenesis and identification of patients with ICI at risk of developing an irAE.
JAK inhibitors may expand the, thus, far limited therapeutic armamentarium to cope with severe, refractory and/or chronic rheumatic irAEs.
Introduction
Immune checkpoint inhibition (ICI) therapies that prevent cytotoxic T-lymphocyte-associated Protein 4 (CTLA-4) and programmed cell death protein 1 (PD-1) from blocking T cell activation are a milestone in cancer management. Their initial success in patients with advanced melanoma and non-small-cell lung cancer (NSCLC) has encouraged their use for other types of solid tumours.1 2 However, the increase in the number of patients under ICI therapy is leading to a rise in the number of patients developing ICI-induced immune-related adverse events (ICI-irAE) resembling chronic autoimmune diseases,3 including rheumatic musculoskeletal and systemic symptoms as well as flares of pre-existing inflammatory diseases.4 De novo arthralgia, inflammatory arthritis, tendinitis/tenosynovitis, enthesitis and (poly-)myalgia have been reported in about 20% of ICI patients in clinical trials, with a large variation in prevalence due to differing criteria and awareness of these side effects.4 5 Intriguingly, the development of ICI-induced irAE has been associated with a better survival and clinical outcome,6–8 including patients with rheumatic irAEs.9–11 However, severe irAEs may force clinicians to terminate ICI therapy due to ICI-irAE-associated mortality for 0.5%–1.5% of patients.12 Fortunately, except for myositis, rheumatic irAEs are seldom fatal but can cause considerable suffering and disability. In contrast to other ICI-irAEs, rheumatic irAEs regularly take a chronic course and require long-term medication.13 While numerous severity-based treatment algorithms for rheumatic irAEs have been formulated to reduce inflammation and patient suffering,4 5 there is an unmet need for evidence-based anti-inflammatory approaches without negative effects for the beneficial antitumor response in this population.9 14–17
In this context, data from our and other groups support the hypothesis that CD8+ T cells (CD8) play an important role in maintaining chronic arthritis and their permanent proinflammatory effector phenotype is fuelled by an enhanced aerobic glycolysis.18–20 While the use of therapies that reduce the CD8 cytotoxic proinflammatory potential such as Janus-kinase inhibitors (JAKi) may be beneficial to control autoimmune arthritis (AA), they might be inappropriate for irAE, since a fully functional CD8 antitumor response is crucial for long-term remission.21 However, CD8 seems to play a role in the induction and/or propagation of irAE since patients with irAE present a clonal expansion of CD8 in the periphery prior to symptom development22 and gene expression profiles of CD8 from patients with irAE are distinct from those who do not develop irAE.8 Nonetheless, functional studies on CD8 in irAE patients that could provide information to evaluate this therapeutic target are largely missing. Therefore, the aim of the present study was to characterise the immunofunctional and metabolic phenotype of the peripheral CD8 pool in patients with rheumatic irAEs and compare these to the CD8-profiles from patients who did not develop irAE under ICI treatment (ICI-CNT), patients with AA (AA-CNT) and patients with AA and a clinical history of malignancy (AA-MAL). We also explored JAK inhibition as a potential therapeutic strategy in ICI-irAEs and AA-MAL by testing whether in vitro blockade of the JAK pathway in the CD8 of these patients results in a major loss of functionality and metabolic remodelling.
Patients and methods
A detailed description of the patient selection and the experimental and statistical methods are found in online supplemental information file 1.
Supplemental material
Results
Patient characteristics
Demographic and clinical data regarding malignancy and autoimmune characteristics are summarised in table 1. Further details on underlying rheumatic diseases, irAEs and malignancies of individual patients are listed in online supplemental table 1. Most ICI patients had a diagnosis of stage III or IV melanoma or NSCLC and all had at least a stable disease as best response. More than half (63.2%) of the patients with ICI-irAE and all in the ICI-CNT group were still under ICI treatment at sample collection. The ICI-CNT group had a shorter disease and ICI treatment duration and higher proportion of men. Musculoskeletal irAEs were verified and treated by a rheumatologist and were characterised by inflammatory arthralgia/arthritis, tenosynovitis and/or polymyalgia, including one patient with an overlap of polymyalgia and suspected mild myositis, another with overlap of spondylarthritis and acute gout, one with concomitant scar sarcoidosis as further irAE, and two with a flare of either pre-existing rheumatoid arthritis (RA) or psoriatic arthritis. Treatment consisted mainly of low-dosed glucocorticoids (GC) ≤10 mg prednisolone-equivalent with only one patient receiving a higher dose at sample collection. Two patients required methotrexate and one received leflunomide for GC sparing, none was previously treated with biologic (b) or targeted synthetic disease-modifying antirheumatic drugs (DMARDs). Three patients showed high disease activity as measured by a disease activity score (DAS28) and five had elevated C-reactive protein (CRP) at sample collection.
Clinical and demographic characteristics of the study participants
The AA-MAL patients had a longer duration and a larger spectrum of malignant diseases though most patients showed complete remission. Most of the AA-MAL group received conventional synthetic (cs) and/or biological disease-modifying antirheumatic drugs (bDMARDs) at sample collection, while GC were used in a lower dosage than for the ICI-irAE group. In contrast to the AA-MAL group, the AA-CNT and AA-JAK groups consisted of patients of younger age, male gender, slightly shorter duration of the rheumatic disease and higher rates of predominantly rheumatoid factor and/or anti-citrulinated protein antibodies (ACPA)-positive RA. CRP levels were low to normal across all AA groups.
Expression of cell-surface markers and release of immune mediators distinguishes the CD8 between patient groups
In vitro culture and T-cell receptor (TCR) stimulation of peripheral blood CD8 for 72 hours did not significantly affect the number of viable cells when compared with ex vivo analysis after cell isolation, and all groups had levels of live cells in excess of 85% (online supplementary figure SF1A). Thus, any subsequent differences observed in marker expression and immune mediator release could not be attributed to general alterations in cell viability. The expression differences in CD8 cell-surface molecules characteristic for activation and effector functions, homing and exhaustion and apoptosis on TCR-mediated stimulation were determined for the total CD8 pool and in its functional subsets defined by the expression of CCR7 and CD45RA. The distribution of naïve, effector (TEMRA), effector memory (TEM) and central memory (TCM) subsets within the total CD8 population was similar for all study groups before culture (Ex vivo: χ2=16.8, p=0.052) and did not change after 72 hours in vitro culture (Nst: χ2=5.007, p=0.83) nor on in vitro TCR-mediated stimulation (St: χ2=9.772, p=0.3692; figure 1A). Significant positive fold changes from baseline in the frequency and expression of activation-related molecules (CD69 and CD25) and homing molecules (CD11a and CD49a) were observed in all CD8 subsets and the total CD8-pool in all groups. Additionally, CD69+ and CD25+ CD8 were significantly more enriched in the ICI-CNT total CD8-pool—but not in any particular subset—than in the ICI-irAE total CD8-pool (figure 1B–D and online supplemental figure SF1C). CD25 expression was higher on the surface of CD8 subsets and the total pool from ICI patients when compared with the ICI-irAE. A few other significant differences in the expression of cell-surface molecules were observed after TCR stimulation of AA-CNT compared with AA-MAL or ICI-irAE and between ICI-CNT and ICI-irAE.
Supplemental material

Immunophenotype and release of immune mediators is different between ICI-irAE and ICI-CNT CD8. (A) Representative overlay dot-plots of CD45RA versus CCR7 expression in unstimulated and TCR-stimulated CD8 and stacked-column graphs showing the distribution of the four main functional CD8 subsets based on CD45RA versus CCR7 expression (naïve: CD45RA+CCR7+; TEMRA: CD45RA+CCR7-; TEM: CD45RA-CCR7-; and TCM: CD45RA-CCR7+) within each patient group. (B) Bar graphs showing the fold-change expression (MFI) of the different markers in the main functional CD8 subsets after TCR-mediated stimulation. (C–E) Bar graphs showing the fold-change in surface-marker frequency (C), surface marker expression (D) and cytokines, and cytotoxic molecules release (E) after TCR-mediated stimulation. (F) Volcano plots showing the differentially expressed molecules between the different patient groups. The horizontal dotted line represents p value<0.05; the horizontal dashed line represents adjusted p value<0.05. For all panels: AA-CNT n=18; AA-MAL n=16; ICI-irAE n=19; ICI-CNT n=10. Representative patients for panel A: #17 AA-CNT list; #14 from AA-MAL list; #16 from ICI-irAE list; and #10 from ICI-CNT list. AA-CNT, autoimmune arthritis; AA-JAKi, autoimmune arthritis Janus-kinase inhibitor; AA-MAL, autoimmune arthritis malignancy; GC, glucocorticoids; ICI-irAE, immune checkpoint inhibitor-immune-related adverse event; NSCLC, non-small-cell lung cancer; PsA, psoriatic arthritis; RA, rheumatoid arthritis; SpA, spondylarthritis.
Total CD8 increased the release of cytotoxic mediators and cytokines after TCR-mediated stimulation (figure 1E) across all groups. However, ICI-CNT CD8 overall presented a more robust secretion of immune mediators and, in particular, the release of cytolytic molecules perforin, granulysin and granzymes A and B higher than for ICI-irAE.
Next, we analysed whether these differences could distinguish AA-CNT from the AA-MAL and ICI-irAE groups (figure 1H). A higher expression of Granzyme A and PD-1 was characteristic for AA-MAL and ICI-irAE CD8 in comparison to AA-CNT, even though only Granzyme A reached a significant adjusted p value for irAE versus AA-CNT. Further molecules that separated these groups from AA-CNT were the cytolytic molecules sFasL and Granulysin (AA-MAL) and CTLA-4 (ICI-irAE). We observed that ICI-irAE CD8 were distinguished from ICI-CNT by a lower expression of activation and homing molecules, proinflammatory cytokines (IFN-γ, TNF-α) and several cytolytic mediators. When searching for molecules that were distinct between ICI-CNT and AA-CNT, we found that they mostly overlapped with the ones separating ICI-CNT from ICI-irAE. However, in both cases, only Perforin reached a significant adjusted p value.
To determine whether clinical or demographic characteristics could contribute to and explain any of the described differences, we correlated the continuous clinical (including treatment modalities) and demographic variables with the experimental data for both the whole study cohort and for each patient group. However, there were no clinical or demographic variables with a significant correlation with the CD8 phenotype across all study groups (online supplemental figure SF2). Moreover, comparison of anti-PD-1-monotherapy and combined anti-PD-1 and anti-CTLA-4 treatment within the ICI-CNT and ICI-irAE patient groups did not yield any significant differences in the CD8 subsets, expression of functional surface molecules or production of cytolytic mediators (online supplemental tables S2 and S3). Furthermore, these parameters also did not differ between ICI-irAE patients with continued versus stopped ICI therapy due to severe irAE in any organ prior to sample collection (online supplemental table S4).
Supplemental material
Metabolic phenotype of CD8
Since the cell-culture media contained (U-13C)-glucose, the in vitro glucose consumption and de novo (U-13C)-lactate production could be precisely quantified by 1H-NMR (figure 2A). CD8 from AA-CNT increased glycolysis on in vitro TCR-stimulation, characterised by a strong de novo (U-13C)-lactate synthesis, which accounted for more than 60% of the total lactate pool. A similar behaviour was also observed for ICI-CNT CD8 (figure 2B and C). Interestingly, the unstimulated ICI-irAE CD8 had a significantly higher de novo (U-13C)-lactate synthesis and a larger contribution of (U-13C)-lactate to the total lactate pool than did the AA-CNT or ICI-CNT CD8. However, (U-13C) lactate levels after TCR-mediated stimulation were comparable between all groups. The oxidative phosphorylation (OXPHOS) rate in all groups bar ICI-irAE dropped after stimulation (Figure 2D). Additionally, we evaluated glucose consumption against expression levels of the glucose transporter 1 (GLUT1) but did not find any significant correlation for any of the groups (data not shown).

irAE CD8 present a Warburg effect-like phenotype when resting and on TCR-stimulation undergo a Crabtree effect-like metabolic shift. (A) Representative 1H NMR sub-spectra of cell-culture media for each group of CD8, either unstimulated or TCR-stimulated. The region covers the [U-12C]-lactate methyl signal and the 13C satellite at higher frequency arising from [U-13C]-lactate. Each spectrum has been normalised separately to its [U-12C]-lactate methyl signal. (B–D) The concentration of [U-13C]-lactate in the cell-culture medium (B), [U-13C]-lactate enrichment (C) and OXPHOS-rate (D) before and after TCR–mediated stimulation for each group. Results are shown as box plots. Each box represents the 25th to 75th percentiles. Lines inside the boxes represent the median. Lines outside the boxes represent the 10th and 90th percentiles. Dots represent outliers. For all panels, AA-CNT n=18; AA-MAL n=16; ICI-irAE n=19; and ICI-CNT n=10. (E) Representative microscopy images of unstimulated ICI-irAE and ICI-CN CD8. (F) Total ATP produced by in vitro cultured CD8 without stimulation or with TCR-mediated stimulation, quantified by measuring the relative ATP-Red fluorescence. Each box represents the 25th to 75th percentiles of nine technical replicates for each patient (AA-CNT n=6; AA-MAL n=6; ICI-irAE n=7; and ICI-CNT n=3). Lines inside the boxes represent the median, lines outside the boxes represent the 10th and 90th percentiles, and dots represent outliers. (H) The correlation between [U-13C]-lactate production and cytokines/cytotoxic molecules release on TCR-mediated stimulation. Numbers show correlations with Spearman R>|0.3|, bold numbers represent p<0.05. AA-CNT, autoimmune arthritis; AA-JAKi, autoimmune arthritis Janus-kinase inhibitor; AA-MAL, autoimmune arthritis malignancy; GC, glucocorticoids; ICI-irAE, immune checkpoint inhibitor-immune-related adverse event; TCR, T-cell receptor.
To confirm that differences in de novo (U-13C)-lactate synthesis and OXPHOS rate represented a preference for ATP-production through aerobic glycolysis, we calculated the percentage of cytoplasmatic ATP within the total cellular ATP (cytoplasmatic plus mitochondrial) by fluorescence microscopy (figure 2E). Without TCR-mediated stimulation, total ATP was significantly higher in ICI-irAE CD8 than in any other group (figure 2F) and directly correlated with an increase in (U-13C)-lactate-enrichment (Spearman R=0.821, p=0.034). In unstimulated cells, cytoplasmatic ATP was the major contributor to the total ATP-pool for all groups (figure 2G). TCR-mediated stimulation did not significantly change the contribution of cytoplasmatic ATP to the total ATP-pool in AA-CNT and ICI-irAE CD8, whereas AA-MAL and ICI-CNT CD8 obtained most of their ATP from the mitochondria.
The release of proinflammatory cytokines and cytolytic molecules positively correlated with increasing (U-13C)-lactate concentrations, particularly the ICI-CNT CD8 (figure 2H). We did not find any general or group-specific correlation between clinical or demographic variables and GLUT1 expression or (U-13C)-lactate production (online supplemental figure SF2).
Different baseline gene-expression profiles distinguish CD8 from ICI patients who develop musculoskeletal irAE
We retrieved the EGAS00001004081 gene expression data8 obtained from peripheral CD8 isolated before the onset of ICI therapy and compared the profiles of patients who later developed specifically arthritis as a rheumatic irAE (13.5%) with those who did not develop any ICI-induced irAE (online supplementary table ST5). Twenty-two transcripts had a significant differential expression between the group of patients who developed an arthritis-irAE and those who did not, and pathway analysis revealed an enrichment of genes involved in cell-population proliferation, immune system development and response to TNF in the group that remained irAE free (figure 3A; online supplementary table ST6). Even though we did not find any significant differences in CD8 phenotype and immune-mediator production among patients with ICI receiving only anti-PD-1 or combined anti-CTLA-4 and anti-PD-1 therapy, it is well documented that patients receiving combined ICI therapy are more prone to develop ICI-induced irAEs in any organ, including arthritis.23 24 Therefore, we conducted a subanalysis of the differences in gene expression at baseline between patients who later developed an arthritis-irAE and those who did not base on the type of ICI therapy. Before ICI treatment with only anti-PD-1, we identified 47 significant transcripts and an enrichment of pathways linked to ATP metabolism and immune response which were differentially expressed in those patients who later developed arthritis-irAE (figure 3B; online supplemental table ST7). No major significant gene-expression differences were observed within the patients who received a combination of anti-CTLA-4 and anti-PD-1 (online supplemental table ST8). Based on arthritis severity, although we identified 18 transcripts that had a significantly different expression (figure 3C, online supplemental table ST9), we could not define any significantly altered pathway. The data did not indicate which patients would require GC to curb arthritis-irAE (online supplemental table ST10). All these data indicate that CD8 differ even before the initiation of ICI.
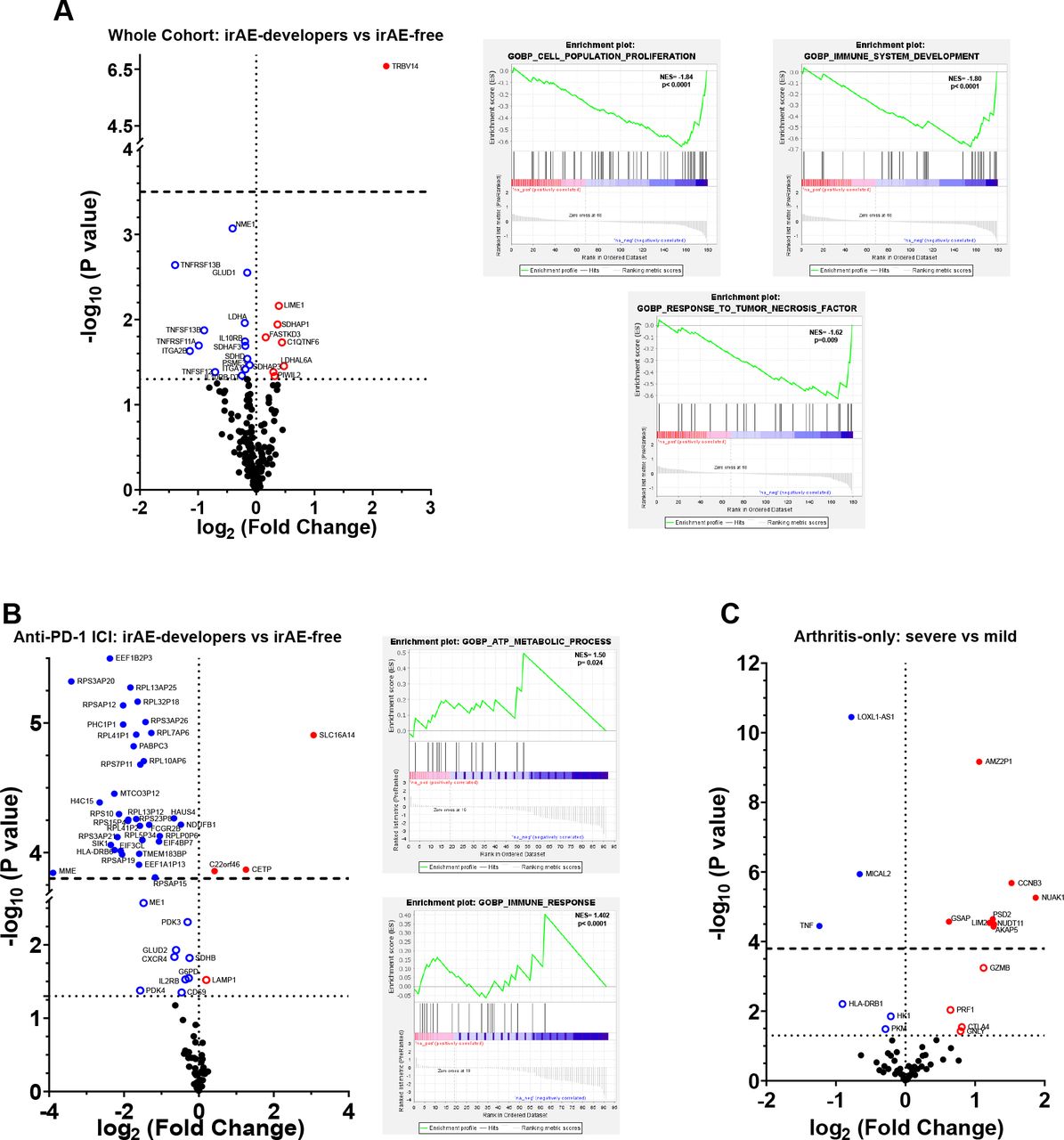
Before therapy begins, patients who later on develop ICI-induced irAE have a different gene expression than those who remain irAE-free. Volcano plots and pathway enrichment plots showing the gene-expression differences before ICI-therapy. (A) Total ICI-treated patients. Those who developed arthritis irAE (n=21) versus those who remained irAE-free (n=135). (B) Patients treated only with anti-PD-1 ICI. Those who developed arthritis irAE (n=7) versus those who remained irAE-free (n=73). (C) ICI-treated irAE patients who developed severe arthritis (grade 3–4; n=7) versus those who developed mild arthritis (grade 1–2; n=14). Horizontal dotted line represents p<0.05; horizontal dashed line represents adjusted p<0.05. ICI, immune checkpoint inhibitor; irAE, immune-related adverse event.
In vitro inhibition of JAK-signalling pathway and TNF-α blockade does not induce major functional or metabolic changes in CD8
To evaluate the effect of JAKi, TCR-stimulated CD8 of all groups were compared with in vitro JAKi-treated CD8. Additionally, TCR-stimulated AA-CNTnbD CD8 were compared with in vitro TCR-stimulated CD8 from AA patients under in vivo JAKi therapy (AA-JAK group). Furthermore, since TNF-α blockade with infliximab is part of the current standard-of-care therapies for ICI-induced irAE, we ran parallel experiments with TNFi-treated CD8, plus compared in vitro TCR-stimulated AA-CNTnbD CD8 to in vitro TCR-stimulated CD8 from patients under in vivo TNFi therapy (AA-TNF group).
In vitro JAKi and TNFi of TCR-mediated stimulation led to a generalised reduction in the expression of most cell surface markers in all patient groups (figures 4A–B and 5A–B). The presence of JAKi significantly reduced the expression of surface HLA-DR in ICI-irAE and ICI-CNT CD8 and of CD11a in AA-MAL and ICI-CNT CD8. TNFi had a significant effect on CD95 expression in ICI-CNT and CD107a and CD57 in AA-nbDCNT CD8. A tendency for a decrease in the frequency of CD8 expressing activation and homing markers was observed for all groups, though not reaching statistical significance for any of the inhibitors (figures 4C and 5C). Since tofacitinib inhibits intracellular signal transduction of several cytokines by blocking JAK1 and JAK3,25 while infliximab only blocks the binding of soluble or transmembrane TNF-α to its receptor,26 we assessed whether in vitro they influenced the release of cytokines and immune mediators by TCR-stimulated CD8 (figures 4D and 5D). Probably due to the focused inhibition of TNF-α-signalling by infliximab, the release of most cytokines and cytotoxic molecules remained unchanged for all groups, and only the ICI-CNT group presented a reduced release of cytolytic mediators. In contrast, in vitro JAKi treatment led to a generalised reduction in the concentration of soluble mediators in all groups, but only ICI-CNT CD8 released significantly less cytolytic mediators in the presence of JAKi. When comparing AA-nbDCNT CD8 to those from AA-JAK or AA-TNF patients after TCR-stimulation (online supplemental figure 3A), we observed a significant reduction in the pool of CD69+ and/or CD25+ in patients under in vivo JAKi or TNFi therapy. Reminiscent of the in vitro behaviour observed for the AA-nbDCNT CD8 under JAKi or TNFi, there was a reduction in the frequency of CD8 expressing homing markers (CXCR4, CD11a and CD49a) within the CD8 pool from AA-JAK and AA-TNF patients, though not significantly different from AA-nbD. No differences were observed in the capacity of TCR-stimulated AA-JAK and AA-TNF CD8 to release cytokines and cytotoxic molecules when compared with their AA-nbDCNT counterparts (online supplemental figure 3B).
Supplemental material

In vitro JAK-pathway inhibition with tofacitinib does not alter the immuno-metabolic profile of ICI-irAE CD8. (A) Representative histograms of changes in the expression of cell-surface molecules by TCR-stimulated CD8 after in vitro JAKi-treatment (AA-nbDCNT, AA-MAL, ICI-irAE and ICI-CNT) and AA-JAK patients. (B–D) Bar graphs showing the fold-changes in surface-marker expression (B), surface marker frequency (C) and cytokines and cytotoxic molecules release (D) of TCR-stimulated CD8 after in vitro JAKi treatment. *p<0.05, **p<0.01 changes between stimulated and JAKi conditions. (E) Inhibition of H838 growth by conditioned media from CD8 (unstimulated, and TCR-stimulated with and without JAKi or TNFi treatment) after 5 days. *p<0.05, **p<0.01; ***p<0.001, ****p<0.0001 between conditioned media versus H838 in medium only. (F) Correlations between H838 cell growth and the concentration of cytokines or cytotoxic molecules in the conditioned cell-culture media. Numbers show correlations with Spearman R>|0.35| and p<0.05. (G–I) Fold change relative to baseline in the concentration of [U-13C]-lactate in the cell-culture medium (G), [U-13C]-lactate enrichment (H) and OXPHOS rate (I) in TCR-stimulated CD8 with (solid symbols) or without (open symbols) in vitro JAKi treatment (AA-MAL, ICI-irAE, and ICI-CNT) or between AA-CNT and AA-JAK patients. *p<0.05, **p<0.01, ***p<0.001 between JAKi-treated and untreated cells. For (B–I): AA-nbDCNT n=10; AA-MAL n=16; ICI-irAE n=19; and ICI-CNT n=10. Representative patients for (A): AA-nbDCNT patient #17 from AA-CNT list; #16 from AA-JAK list; #14 from AA-MAL list; #16 from ICI-irAE list; and #10 from ICI-CNT list. AA-JAKi, autoimmune arthritis Janus-kinase inhibitor; AA-MAL, autoimmune arthritis malignancy; ICI-irAE, immune checkpoint inhibitor-immune-related adverse event; TCR, T-cell receptor.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In vitro TNF-α inhibition with infliximab does not alter the immuno-metabolic profile of ICI-irAE CD8. (A) Representative histograms of changes in the expression of cell-surface molecules by TCR-stimulated CD8 after in vitro TNFi-treatment (AA-nbDCNT, AA-MAL, ICI-irAE and ICI-CNT) and AA-TNF patients. (B–D) Bar graphs showing the fold-changes in surface-marker expression (B), surface marker frequency (C) and cytokines and cytotoxic molecules release (D) of TCR-stimulated CD8 after in vitro TNFi treatment. *p<0.05, **p<0.01 changes between stimulated and TNFi conditions. (E–G) Fold change relative to baseline in the concentration of [U-13C]-lactate in the cell-culture medium (G), [U-13C]-lactate enrichment (H) and OXPHOS rate (I) in TCR-stimulated CD8 with (solid symbols) or without (open symbols) in vitro TNFi treatment (AA-MAL, ICI-irAE and ICI-CNT) or between AA-CNT and AA-TNF patients. *p<0.05 between TNFi-treated and untreated cells. For (B–G): AA-nbDCNT n=4; AA-MAL n=6; ICI-irAE n=7; and ICI-CNT n=3. Representative patients in (A): AA-nbDCNT patient #17 from AA-CNT list; AA-TNF patient #13 from AA-CNT list; #14 from AA-MAL list; #16 from ICI-irAE list; and #10 from ICI-CNT list. AA-JAKi, autoimmune arthritis Janus-kinase inhibitor; AA-MAL, autoimmune arthritis malignancy; ICI-irAE, immune checkpoint inhibitor-immune-related adverse event.
The in vitro expansion of H838 cells could be inhibited for 5 days using conditioned medium from TCR-stimulated CD8 but not with medium containing only JAKi or TNFi (figure 4E; online supplemental figure SF3C-E). The conditioned media from AA-nbDCNT, AA-TNF and AA-JAK patients’ CD8 presented the strongest inhibitory effect. In contrast to ICI-CNT and AA-MAL, conditioned media from ICI-irAE CD8 cultured in the presence of JAKi had comparable inhibitory capacity on H838 cell growth to media without JAKi. In vitro TNFi-treated media from all groups were able to inhibit H838 expansion. Significant negative correlations between H838 inhibition and the concentration of cytokines or cytolytic molecules were evident in the CD8-conditioned media from AA-CNTnbD, AA-JAK and ICI-CNT patients (figure 4F), but none was observed for AA-TNF or in vitro TNFi (data not shown).
The metabolic profile of CD8 of AA-nbDCNT was generally more glycolytic than in AA-JAK and AA-TNF patients with higher (U-13C)-lactate production and lower OXPHOS rate (figures 4G–I and 5E–G and online supplemental figure 3F). Except for a lower enrichment of (U-13C)-lactate in AA-MAL CD8 under in vitro JAKi treatment, no other TNFi-induced or JAKi-induced changes were observed in the metabolic profile of ICI-irAE, ICI-CNT or AA-MAL CD8.
Discussion
Functional and phenotypical changes in CD8 have been generally associated with the success of antitumor response and are, thus, the core of ICI therapy.27 28 However, such changes are equally contributing to the pathophysiology of chronic AA.19 20 29 This poses a major challenge in the treatment of ICI-induced arthritis as inhibition of CD8 would be required for sustained arthritis therapy, which would, however, limit the antitumor effects. To clarify some of these aspects, we have compared phenotype, functional and metabolic changes in peripheral CD8 from ICI patients who either developed or did not develop musculoskeletal irAEs with those from AA patients and tested the effects of in vitro JAKi and TNFi treatment on CD8 associated with antitumor response. The fraction of cells expressing effector/activation and homing markers within the total CD8 pool and the different CD8 subsets, as well as the amount of released immune mediators, was similar between patients with AA-CNT and ICI-irAE, but lower than was observed for ICI-CNT. This AA-like profile was independent of arthritis symptom duration and remained in those ICI-irAE patients who had stopped ICI therapy. Thus, it suggests that ICI-induced arthritis imprints a lasting phenotype on peripheral CD8. Alterations in the phenotype of peripheral blood CD8 have equally been reported in the blood of thymic epithelial tumour and metastatic patients with NSCLC developing different forms of irAEs,30 in the epidermis of melanoma, renal cell carcinoma, gastric cancer and lung cancer patients with ICI-induced psoriasis-like dermatitis31 and in the colon epithelium of melanoma patients with ICI-induced colitis.32
Metabolic remodelling from OXPHOS towards aerobic glycolysis is a hallmark of CD8 activation,33–35 as this allows cells to produce ATP much faster than through OXPHOS.36 Patients with chronic AA display a permanent and exacerbated lactate production (Warburg effect), which is associated with a glycolytic profile and lower OXPHOS that maintains chronic cytotoxicity. On TCR stimulation, CD8 from AA patients further drop their OXPHOS rate and rely solely on aerobic glycolysis19 in a process known as the Crabtree effect. Thus, it was not surprising to observe that unstimulated ICI-irAE CD8 were able to release large amounts of newly synthesised lactate, which correlated to a higher amount of total ATP. Moreover, and like what was observed in the AA-CNT CD8 but unlike ICI-CNT CD8, TCR stimulation might have triggered a Crabtree effect-like profile in ICI-irAE CD8 for they maintained glycolysis as their major source of ATP. Since resting ICI-irAE CD8 were more glycolytic than ICI-CNT but released less cytotoxic and proinflammatory mediators, it is possible that ICI-irAE CD8 have a more robust biosynthetic balance to sustain their effector/antitumor functions for longer periods when compared with ICI-CNT and might contribute to the better clinical outcomes observed in patients with ICI-irAE. However, the downside of keeping a steady proinflammatory and cytotoxic effector phenotype for longer periods is that it may render ICI-irAE CD8 with a RA-like profile, which favours the surge and relapse of irAE and may contribute to the chronic course of rheumatic irAEs.13
Gene-expression analysis has shown that the development of different irAEs has been associated with pre-ICH and post-ICI downregulation of CXCR1 on peripheral CD8 in melanoma patients.8 Here, we reanalysed the same pre-ICI gene-expression data set focusing on those patients who developed specifically arthritis as a rheumatic irAE. Even though the number of available samples was sparse—which limits data interpretation—the results suggest a baseline impairment in the upregulation of TNF-signalling and proliferation pathways. These differences appear to remain after arthritis-irAE has developed, since CD8 from ICI-irAE patients released less TNF and expressed less CD25 than those from ICI-CNT. Collectively, these are relevant findings in the context of the lively discussion on the beneficial or detrimental effects of TNF inhibition as a treatment option for ICI patients with severe irAEs.37–39 Since anti-PD-(L)1- rather than anti-CTLA-4-monotherapy is associated with a higher incidence of rheumatic irAEs,24 40 it justified a separate gene-expression analysis comparing ICI patients who received only anti-PD-1 therapy. It was interesting to observe already at baseline the enrichment of the ATP metabolism-related pathway in those CD8 from patients who later developed arthritis irAE and which could be mirrored by the data obtained for ICI-irAE CD8 from patients with established arthritis. Therefore, this suggests that even before ICI therapy, CD8 from patients who later develop arthritis irAE, present a gene-expression profile that already indicates a different immunometabolic phenotype than those that remain irAE free.
Currently, therapeutic algorithms for irAE-arthritis rely on the defensive use of GC, csDMARDs and TNF- or IL-6-blockers.4 5 However, the use of TNF-inhibition in irAEs is increasingly controversial37–39 and our data suggest that CD8 from ICI-irAE patients present a downregulation of the TNF-signalling pathway and release less TNF than ICI-CNT. Thus, finding other therapeutic strategies to curb ICI-induced musculoskeletal inflammation and maintain antitumor activity will be required to meet current clinical needs in the management of ICI-irAEs. Preclinical studies on human cancer cell lines have shown that JAK-pathway inhibition impairs tumour growth.41 42 Still, new data on increased risk of malignancy in patients with RA associated with tofacitinib use put the previously favourable assessment of JAKi in vitro and in vivo into question.43 44 Clinicians remain hesitant in general to use JAKi in patients with a history of malignancy due to the shorter observational time in premarketing and postmarketing studies compared with most bDMARDs and, in line with this cautious attitude, only one JAKi-treated patient versus six patients with bDMARD therapy were present in our AA-MAL cohort. However, considering the limited treatment options, one needs to expand the therapeutic armamentarium to cope with severe and/or chronic ICI-irAEs, the latter being a frequent course of rheumatic irAEs.13 Therefore, the beneficial effects, as well as potential risks of JAKi in musculoskeletal and other irAEs, should be further investigated, particularly when keeping in mind the increasing number of available JAKi with minor, but clinically relevant, differences in their modes of action. Of note, tofacitinib was previously successfully used for one ICI patient with arthritis-irAE45 and a case series of GC-refractory myocarditis-irAE.46 In view of this, we carried out in vitro experiments to explore the feasibility of using JAK-pathway inhibition by tofacitinib to control CD8 proinflammatory activity without severely compromising antitumor response and compared the data to TNF-α blockade. Our data suggest that in vitro tofacitinib, similarly to infliximab, did not significantly reduce the release of cytotoxic mediators by ICI-irAE CD8. Due to constraints imposed by the limited volume of collected blood, we could only indirectly measure the in vitro capacity of tofacitinib or infliximab-treated CD8 to inhibit tumour cell growth using conditioned media and not through a direct coculture system. Nevertheless, it seems that. in our experimental setting, the antitumor capacity of CD8-conditioned media from patients with ICI-irAE could be maintained in the presence of both drugs, even if it was lower than observed for the cancer-free AApatients. Additionally, in vitro tofacitinib and infliximab treatment did not reduce aerobic glycolysis, essential for maintaining antitumor functions in CD8.47
The lack of a group of AA-free patients with cancer with ongoing tumour activity and without ICI therapy and the use of only one type of JAK inhibitor for the in vitro studies (a constraint imposed by the reduced number of cells obtained from each patient) are potential limitations of our study. To counter this limitation, we included the patients AA-MAL who had simultaneously a clinical history of malignancy (some still with active tumours) and chronic arthritis, and AA-JAK patients with chronic AA receiving different types and doses of JAK inhibitors. Since the AA-MAL and the ICI-irAE CD8 presented similar profiles, even in their response to in vitro JAKi, we assume that ICI, other cancer therapies, or ongoing tumour activity did not play a major role in the observed immune and metabolic profile changes. Since the CD8 phenotype was quite consistent among all AA-JAK patients, we considered that limiting the in vitro studies to one type of JAKi does not reduce the veracity of our findings.
Further potential limitations are the uneven distribution of anti-PD-1 monotherapy and combination treatment of anti-CTLA-4 and anti-PD-1 between ICI-irAE and ICI-CNT groups as well as the shorter duration of ICI therapy and higher proportion of ongoing treatment in the latter group. This bias is a result of the exclusion criterion of moderate to severe irAEs in any organ in the ICI-CNT and the fact that patients exposed to anti-CTLA-4 or the combination treatment generally show a higher incidence, increased severity and faster onset of irAEs23 24 and, therefore, are less likely to remain irAE free over a longer period of ICI treatment. To address this problem, we compared the expression of cell-surface markers and release of immune mediators between ICI-irAE and ICI-CNT patients with monotherapy and combination treatment and ICI-irAE patients with ongoing and discontinued ICI treatment but did not observe any significant differences. Therefore, we hypothesise that previous anti-CTLA-4 exposure and/or ongoing ICI treatment at sample collection were not the driving factors behind the differences in metabolic and immune-effector profiles between ICI-irAE and ICI-CNT groups.
Overall, our study shows that CD8 from patients with cancer who develop musculoskeletal irAEs during ICI treatment have a distinct immune-effector and metabolic profile from those ICI patients that remain irAE free. The irAE profile is characterised by lower cytotoxic and proinflammatory activity and more aerobic glycolysis and overlaps with the profile observed in AA-CNT and AA-MAL CD8. This suggests that chronic inflammatory arthritis has a unique fingerprint that can be used to direct new therapeutic strategies for managing ICI-induced irAE. One such therapeutic approach may involve JAK pathway inhibition that does not interfere with the antitumor capacity of ICI-irAE CD8 in our experimental model. Thus, future trials on tumor-bearing mice with (poly)arthritis or controlled clinical trials on ICI-irAE patients using JAKi should be the next step to improve therapeutic outcomes while maintaining ICI efficacy together with simultaneous irAE control.
Data availability statement
Data are available upon reasonable request. All data are presented in the manuscript. Raw experimental data will be made available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
The study was officially approved by the institutional ethics committee of the Heidelberg University (ethic votum numbers: S-096/2016, S-391/2018, S-686/2018). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
Benjamin Fairfax and Christine Ye (Department of Oncology, University of Oxford) for granting access to the gene-expression data and help with its analysis by the physicians and nursing teams of the skin cancer ambulatory unit of the National Center for Tumor Diseases, Heidelberg University Hospital; and the nursing team of the blood collection unit of the Medical Clinic, Heidelberg University Hospital. All microscopy was executed at the ZMBH Imaging Facility.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
KB and FVK contributed equally.
Contributors KB, JG, LD, TS, JCH, PC and H-ML patient selection and recruitment; FVK, RAC, HL, CHH and MS-C experimental design; FVK, KDK, HL, CHH, JG, MS-C data collection; KB, FVK, RAC, HL, KDK, MS-C data analysis; KB, FVK, RAC, HL, LD, JG, KDK, PC, JCH, H-ML and MMSC manuscript writing and revision; KB, H-ML and MS-C funding. MMSC guarantor for the overall content.
Funding This work was funded by the unrestricted investigator-initiated grant WI245418 from Pfizer Pharma GmbH to MMSC. The funding body did not have any influence on the study design, nor on the data acquisition and analysis and interpretation. FV Kraus was supported by an Add-On Fellowship of the Joachim Herz Foundation.
Competing interests KB: Consultancy and/or speaker fees and/or travel reimbursements: Abbvie, Bristol Myers Squibb (BMS), Gilead/Galapagos, Janssen, Merck Sharp & Dohme (MSD), Mundipharma, Novartis, Pfizer, Roche, Viatris, UCB. Scientific support: Medical Faculty of University of Heidelberg, Rheumaliga Baden-Württemberg e.V., AbbVie, Novartis. MMSC: Scientific support: Novartis, Pfizer. JCH: honoraria: BMS, MSD, Novartis, Roche, Pierre Fabre, Sanofi, Almirall; consultant or advisory role: MSD, Pierre Fabre, Sunpharma; Scientific support: BMS; Travel support: Pierre Fabre. PC: honoraria: Roche, Takeda, Gilead, AstraZeneca, Novartis; scientific support: Roche, Takeda, Amgen, Merck, AstraZeneca, Novartis; travel support: AstraZeneca, Merck, Janssen, Daiichi Sankyo, Takeda, Novartis, Elli Lilly; data safety monitoring and/or advisory board: Pfizer, Chugai, Boehringer Ingelheim, Roche. JG: honoraria: Galapagos; travel support: Elli Lilly. HML: Scientific funding: Abbvie, Novartis, Pfizer, Roche; consulting fees and honoraria: Abbvie, AstraZeneca, Actelion, Amgen, Bayer Vital, Boehringer Ingelheim, BMS, Celgene, GlaxoSmithKline (GSK), Galapagos, Janssen, Elli Lilly, Medac, MSD, Novartis, Pfizer, Roche, Sanofi, UCB; travel support: Abbvie, AstraZeneca, Boehriner Ingelheim, BMS, Celgene, GSK, Gilead, Janssen, Elli Lilly, MSD, Novartis, Pfizer, Roche, Sanofi, UCB; data safety monitoring and/or advisory board: Abbvie, AstraZeneca, Amgen, Boehriner Ingelheim, BMS, Celgene, GSK, Gilead, Janssen, Elli Lilly, Medac, MSD, Novartis, Pfizer, Roche, Sanofi, UCB
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.