Article Text

Abstract
Objective To determine contributions and functions of autoantibodies (Abs) directed to the angiotensin receptor type 1 (AT1R), which are suggested to be involved in the pathogenesis of AT1R Abs-related diseases such as systemic sclerosis (SSc).
Methods C57BL/6J mice were immunised with membrane-embedded human AT1R or empty membrane as control. Mice deficient for CD4+ or CD8+ T cells and B cells were immunised with membrane-embedded AT1R or an AT1R peptide proposed to be a dominant T cell epitope. A monoclonal (m)AT1R Ab was generated by hybridoma technique and transferred into C57BL/6J and AT1Ra/b knockout mice. The induced phenotype was examined by histology, immunohistochemistry, immunofluorescence, apoptosis assay and ELISA. In vitro, Abs responses towards AT1R were measured in cells of different origins and species.
Results AT1R-immunised mice developed perivascular skin and lung inflammation, lymphocytic alveolitis, weak lung endothelial apoptosis and skin fibrosis accompanied by Smad2/3 signalling, not present in controls or mice deficient for CD4+ T and B cells. The AT1R peptide 149–172 provoked lung inflammation. Application of the mAT1R Ab induced skin and lung inflammation, not observed in AT1Ra/b knockout mice. In vitro, AT1R Abs activated rat cardiomyocytes and human monocytes, enhanced angiotensin II-mediated AT1R activation in AT1R-transfected HEK293 cells via AT1R binding and mAT1R Ab-activated monocytes mediated the induction of profibrotic markers in dermal fibroblasts.
Conclusion Our immunisation strategy successfully induced AT1R Abs, contributing to inflammation and, possibly, to fibrosis via activation of AT1R. Therefore, AT1R Abs are valuable targets for future therapies of SSc and other AT1R Ab-related diseases.
- autoantibodies
- scleroderma, systemic
- inflammation
- immune system diseases
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Angiotensin receptor type 1 (AT1R) autoantibodies (Abs) are suggested to contribute to pathologies found in systemic sclerosis as well as in other conditions with high values of AT1R Abs.
What does this study add?
AT1R Abs have the potential to induce lung and skin inflammation, dermal fibrosis and endothelial apoptosis.
Our immunisation strategy to induce functionally active Abs can serve as model to generate Abs against other complex transmembrane proteins.
AT1R Abs can act agonistic and allosteric in combination with the orthosteric ligand angiotensin II.
How might this impact on clinical practice or future developments?
As an important ligand of AT1R, AT1R Abs are a novel target for future therapies in diseases associated with high values of AT1R Abs.
Introduction
Angiotensin receptor type 1 (AT1R, AGTR1) is a G protein-coupled receptor (GPCR) centrally involved in the regulation of vascular tone, proliferation of vascular smooth muscle cells, extracellular matrix generation and inflammation.1–3 AT1R binds its endogenous ligand angiotensin II (Ang II) in a saturable manner with high structural specificity and affinity, thereby mediating most of the physiological actions of Ang II.4 5 Physiologically, AT1R is expressed in vascular tissues, including skin and lung.1 6–8 Human and the two subtypes of rodent AT1R, AT1Ra and AT1Rb, respectively, share a high degree of homology.9 Notably, increased levels of circulating autoantibodies targeting AT1R (AT1R Abs), partially accompanied by functional activity, have been found in patients with renal transplant rejection, glomerulosclerosis, preeclampsia and autoimmune diseases such as systemic sclerosis (SSc), a severe inflammatory, vascular and fibrotic disease.10–14 In the latter, high serum levels of AT1R Abs predicted mortality, pulmonary arterial hypertension and digital ulcers.11 15 16 In vitro, SSc patient-derived IgG containing high levels of AT1R Abs induced gene expression of transforming growth factor ß (TGFβ), expression of adhesion molecules and chemokines in endothelial cells, release of interleukin 8 (IL-8) and CC-chemokine ligand 18 (CCL18) in leukocytes and production of collagen type I in fibroblasts.11 17–19 Blockade by AT1R antagonists indicated specificity for AT1R.17 18 However, no in vivo model exists, demonstrating appearance of SSc manifestations by induction of high AT1R Ab levels. Thus, the study attempted to find out if immunisation of C57BL/6J mice with membrane extracts (ME) of cells overexpressing human AT1R induces AT1R Abs, pathologies and the relevance of T and B cells. Further, a monoclonal (m)AT1R Ab as well as AT1Ra/b knockout mice were applied to validate AT1R Ab binding in vivo.20 Finally, rodent and human AT1R-expressing cells were stimulated in vitro with the mAT1R Ab or murine IgG containing AT1R Abs. AT1R Ab binding and activation of AT1R were measured by label-free dynamic mass redistribution (DMR) technology in AT1R-transfected HEK293 cells.21 22 Altogether, AT1R immunisation of C57BL/6J mice led to interstitial lung disease (ILD) and skin fibrosis, accompanied by high levels of AT1R Abs, whose generation most likely depends on CD4+ T cells and B cells. The local application of the mAT1R Ab also led to signs of skin and lung inflammation in C57BL/6J mice, which were diminished in the AT1Ra/b knockout mice. Further, the in vitro results suggest that interactions between rodent or human AT1R-expressing cells and murine IgG containing AT1R Abs or the mAT1R Ab, either alone or in combination with Ang II, promote activation of AT1R and/or subsequent functional responses.
Materials and methods
Detailed experimental procedures are reported in the online supplemental materials and methods (see online supplemental materials and methods).
Supplemental material
Results
Immunisation with AT1R induces pulmonary disease and AT1R Abs in vivo
Following two immunisations with AT1R ME, the C57BL/6J mice developed symptoms of ILD as indicated by lymphocytic alveolitis in bronchoalveolar lavage fluid (BALF) which is not observed in the control mice (figure 1A). Histopathological analyses showed a profound inflammation in intra-alveolar areas of the lung, not found in the control mice (figure 1B). Accordingly, the inflammatory score, reflecting number and size of infiltrates in the lung, was higher in the AT1R-immunised mice than in the corresponding controls (figure 1C, p<0.01). Immunohistochemical staining revealed a dominant presence of T and B cells in the lung (figure 1D). Of note, immunisation with AT1R also induced perivascular inflammation in the lung, again mainly consisting of T and B cells (online supplemental figure 1A). Moreover, IgG deposition indicative of an immune reaction was observed in lung tissue of the AT1R-immunised mice and not present in the animals used as controls (figure 1E). The AT1R-immunised mice displayed high levels of circulating AT1R Abs, peaking at day 56 after the first immunisation, and not detected in control mice (figure 1F). The AT1R-reactive Abs belonged to the IgG1, IgG2a and IgG2b subclasses, while the IgG3 subclass was not observed (figure 1G). In addition, the percentage of apoptotic endothelial cells was higher in the AT1R-immunised mice compared with the control mice (online supplemental figure 1B,C, p<0.01). However, vascular remodelling, a key feature of vasculopathy, as well as lung fibrosis was not observed in the AT1R-immunised mice as investigated by histological and biochemical analyses (figure 1H,I).

Immunisationwith AT1R induces signs of lung inflammation. (A) Quantitative analysis of leucocyte populations in bronchoalveolar lavage fluid obtained from controls and hAT1R-immunised mice 9 weeks after the first immunisation. (B) Histology of the lungs of control and hAT1R-immunised mice (200×, scale bar=100 µm). (C) Analysis of lung inflammation quantified by scoring the sizes and numbers of immune infiltrates in intra-alveolar areas in a double-blinded fashion. The results are presented as mean±SD. Statistical analyses were performed using the Mann-Whitney U test (**p<0.01). (D) Cellular composition of the infiltrates in the mouse lungs (200×, scale bar=100 µm) as detected by immunohistochemistry (IHC). Both intra-alveolar and perivascular infiltrations were evaluated. Representative micrographs of H&E staining are shown. (E) Immunofluorescence staining of lung cryosections from hAT1R-immunised mice (630×, scale bar=25 µm) featured murine immunoglobuline G (IgG) deposition (red) and nuclei were stained with 4',6-Diaminodino-2-phenylindol (DAPI, blue). Total anti-AT1R IgG (F) and subclasses of anti-hAT1R IgG (G) were detected by ELISA using plates coated with membrane extracts from CHO cells overexpressing hAT1R and the appropriate detection antibodies. The titre of anti-AT1R antibodies was defined as the dilution at which the optical density (OD) value reached half of the maximal OD values of the curve. The results are presented as mean±SD, and statistical analysis was performed using Student’s t-test (**p<0.01, ***p<0.001). (H) Representative lung sections from control and hAT1R-immunised mice stained with Masson trichrome (100×, scale bar=100 µm). (I) Collagen content in the lungs of control and hAT1R-immunised mice. The collagen content was determined using a Sircol collagen detection kit and is expressed as μg per mg of lung tissue. Data are presented as mean±SD, and statistical analysis was performed using Student’s t-test. AT1R, angiotensin receptor type 1.
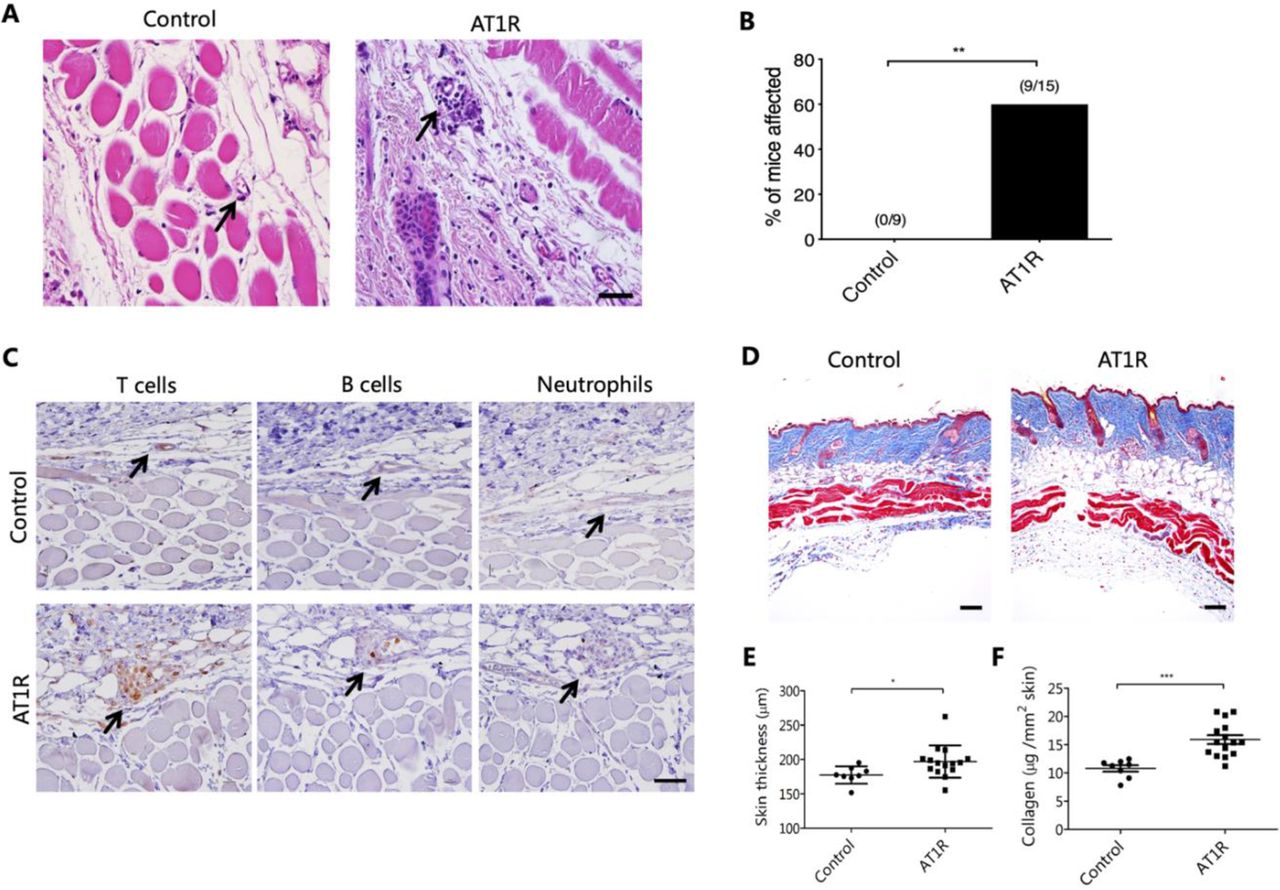
Immunisation with AT1R provokes SSc-like skin inflammation and fibrosis in vivo
At day 63 after the first immunisation, 60% of the AT1R-immunised mice showed dermal perivascular infiltrates, which were not present in the controls (figure 2A,B). These infiltrates were dominated by T cells, while B cells and neutrophils were less prominent or absent (figure 2C). In addition, AT1R immunisation resulted in increased skin thickness, which was not seen in the control mice (figure 2D,E, p<0.05). The signs of skin fibrosis observed histologically were validated by quantitative assessment of skin collagen using Sircol reagent. Our results revealed an increased expression of collagen by 48% in the AT1R-immunised mice compared with the control mice (figure 2F, p<0.001), indicating development of fibrosis in this organ. Further, immunofluorescence staining showed an increased number of alpha-smooth muscle actin (α-SMA)+myofibroblasts and cells positive for Smad2/3 phosphorylation in the AT1R-immunised mice compared with controls (online supplemental figure 2), supporting a role of myofibroblasts and TGFβ signalling in the fibrotic process. However, obliterative vasculopathy was not observed in the AT1R-immunised mice. Furthermore, histological analyses of the kidney, heart, intestine and oesophagus did not show any abnormalities (online supplemental figure 3), indicating that immunisation with AT1R preferentially affects murine lung and skin.

Immunisationwith AT1R induces signs of skin inflammation and fibrosis. (A) Representative H&E staining of skin sections from control and hAT1R-immunised mice (400×, scale bar=50 µm). Black arrows indicate blood vessels in the dermis. (B) Incidence of perivascular inflammatory infiltrates in the skin of control and hAT1R-immunised mice. The numbers on top of the bars show the ratios between the number of mice with perivascular inflammation and the total number of evaluated mice. Statistical analysis was performed using Fisher’s exact test (**p<0.01). (C) Cellular composition of the perivascular infiltrates in the skin of controls vs AT1R-immunised mice (400×, scale bar=50 µm) as detected by IHC. (D) Representative dorsal skin sections after Masson trichrome staining of control or hAT1R-immunised mice (100×, scale bar=100 µm). (E) Quantitative analysis of the thickness of the collagen layer as indicated by the blue area in sections stained with Masson trichrome. (F) Collagen content of the skin in control and hAT1R-immunised mice determined by Sircol collagen detection kit and expressed as μg per mm2 of skin. Data are presented as mean±SD, and statistical analyses were performed using the Mann-Whitney U test or Student’s t-test (*p<0.05, **p < 0.01, ***p<0.001). AT1R, angiotensin receptor type 1.
CD4+ T and B cells are indispensable for lung and skin pathology following immunisation with AT1R
To clarify whether lymphocytes are required for development of AT1R-induced pathologies, mice deficient in CD4+ T cells, CD8+ T cells and B cells were chosen for AT1R immunisation. In the absence of CD4+ T and B cells, mice were unable to generate AT1R Abs in contrast to mice deficient for CD8+ T cells or wild-type controls. Notably, B cell-deficient mice displayed decreased serum IL-6, supporting a role of B cell-derived IL-6 in the inflammatory response towards AT1R immunisation (online supplemental figure 4). Compared with wild-type controls, development of pulmonary inflammation was dramatically reduced in mice deficient for CD4+ T cells and B cells, but not in the absence of CD8+ T cells (figure 3A,B). In line with this, AT1R immunisation induced perivascular skin inflammation in 5 of 7 (71%) wild-type mice, in 50% (4 out of 8 mice) of the CD8+ T cell-deficient mice, but in none of the 11 mice deficient for CD4+ T cells and only in 11.1% (1 out of 9 mice) of the B cell-deficient mice (figure 4A,B). Moreover, AT1R-induced skin thickening and quantitative collagen expression reflecting fibrosis were observed in wild-type and CD8+ cell-deficient mice, but not in CD4+ T cell-deficient or B cell-deficient mice (figure 4C–E). Of note, the number of macrophages in the BALF was not different between the various groups of mice (data not shown). Overall, the data suggest that the generation of AT1R Abs and inflammation require interactions between CD4+ T and B cells. Given the essential role of CD4+ T cells in this novel mouse model, we next investigated the pathogenicity of AT1R-specific CD4+ T cells. By using NetMHCIIpan 3.1 software, AT1R 149–172 peptide was predicted to harbour a potential CD4+ T cell epitope showing strong binding affinity to a murine MHC class II molecule. Immunisation with the AT1R 149–172 peptide induced strong lung inflammation in C57BL/6 mice, while such inflammation was not observed in solvent controls. In addition, no inflammation was observed in skin, heart or kidney in either group of mice (online supplemental figure 5).

CD4+ T and B cells are indispensable for lung pathology following immunisation with AT1R. (A) Representative micrographs of H&E-stained lung sections of wild-type (WT), CD4+ T cell-deficient (CD4 KO), CD8+ T cell-deficient (CD8 KO) and B cell-deficient (muMT) mice immunised with control ME or AT1R-ME. Black arrows indicate inflammatory cell infiltrates in the lung. Scale bar=100 µm. (B) Quantitative analysis of pulmonary inflammation in mice assessed by size and number of infiltrates scored in a double-blinded fashion. Statistical analyses were performed using Mann-Whitney U test or Student’s t-test depending on the normal distribution of data. *p<0.05, ***p<0.001. AT1R, angiotensin receptor type 1.
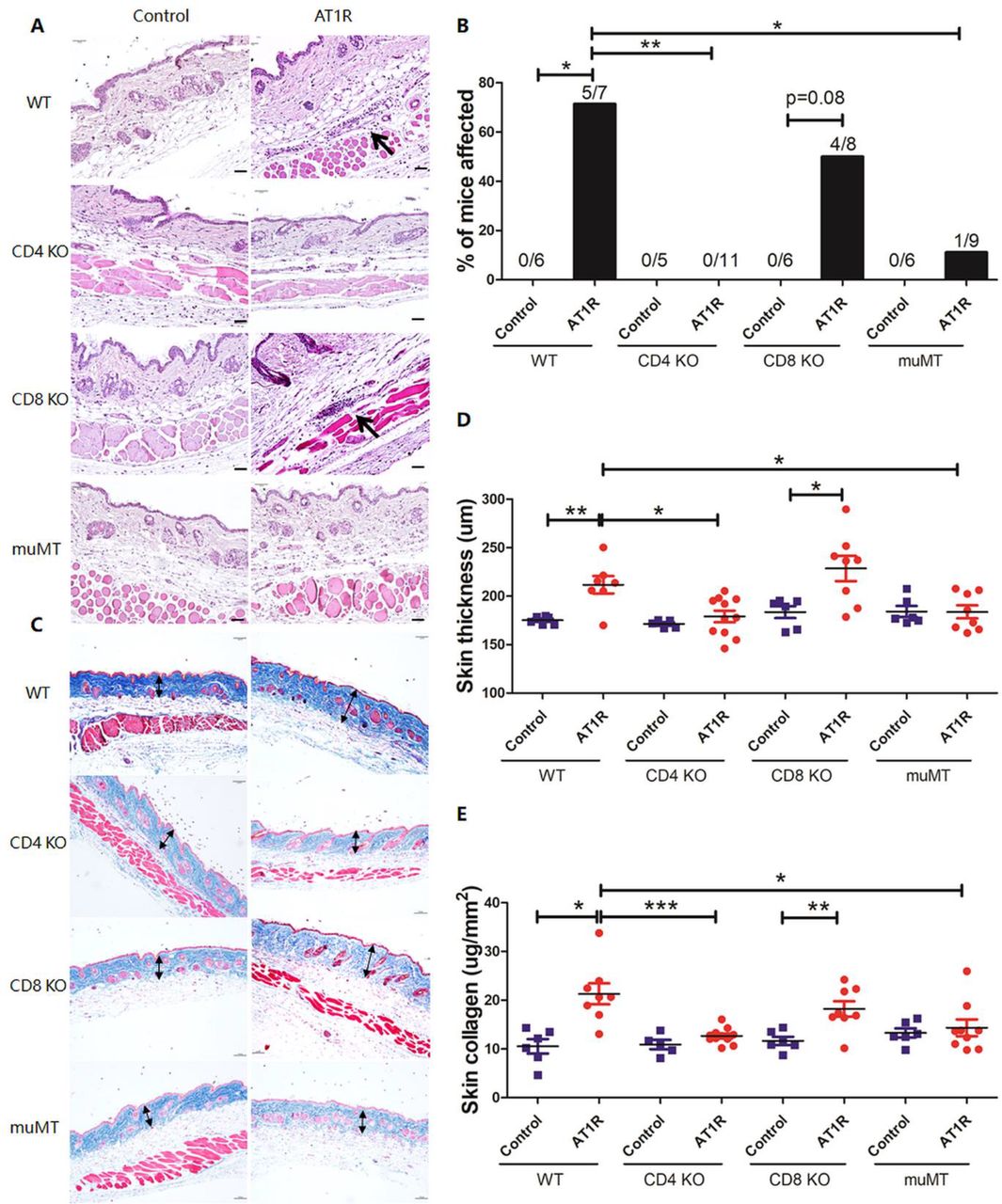
CD4+ T and B cells are indispensable for skin pathology following immunisation with AT1R. (A) Representative micrographs of H&E-stained skin sections of wild-type (WT), CD4+ T cell-deficient (CD4 KO), CD8+ T cell-deficient (CD8 KO) and B cell-deficient (muMT) mice immunised with control ME or AT1R-ME. Black arrows indicate inflammatory cell infiltrates around blood vessels. Scale bar=50 µm. (B) Incidence of skin inflammation as indicated by orange colours. P values were calculated by using Fisher’s exact test (*p<0.05, **p<0.01, ***p<0.001). (C) Representative micrographs of Masson trichrome-stained skin sections. Double-headed arrows indicate the collagen layer of the skin. Scale bar=100 µm. (D) Quantitative analysis of collagen layer thickness stained in blue from the Masson trichrome staining. (E) Quantitative analysis of collagen content determined by Sircol collagen detection kit and expressed as μg per mm2 of the skin. Statistical analysis was performed using Mann-Whitney U test or Student’s t-test depending on the normal distribution of data (*p<0.05, **p<0.01, ***p<0.001). AT1R, angiotensin receptor type 1.
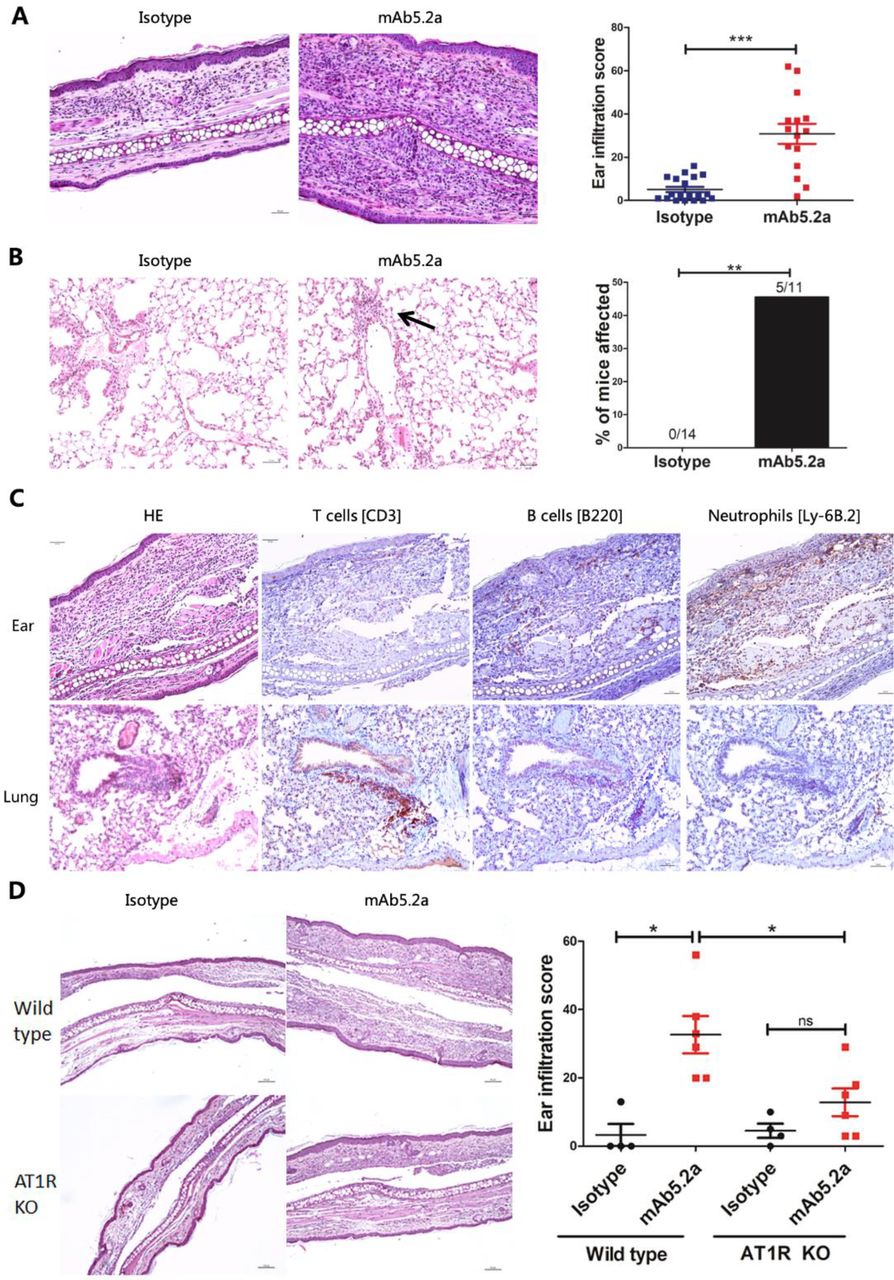
Administration of a monoclonal AT1R Ab induces SSc-like signs of skin and lung inflammation in vivo
A single intradermal injection of IgG derived from the AT1R-immunised mice into the ear of naive C57BL/6J mice induced more inflammation compared with IgG injection derived from control mice (online supplemental figure 6), suggesting a possible role of AT1R Abs in the induction of local inflammation. To examine if AT1R Abs contribute to local inflammation, the monoclonal Ab 5.2a, which exhibits specificity to human AT1R by ELISA and western blot (data not shown) was studied in more detail. Repetitive intradermal injections of this mAT1R Ab into the ears of C57BL/6J mice, but not of the IgG isotype control, induced cellular infiltrations into the skin (figure 5A, p<0.01) as well as perivascular lung inflammation in 5 out of 11 mice, which was not observed in mice treated with IgG isotype control (figure 5B, p<0.01). In the skin, infiltrations consisted of neutrophils and to a much lesser extent of B and T cells, whereas lung infiltrates contained mainly T cells, but no neutrophils (figure 5C). To validate a potential role of an Ab-mediated interaction with AT1R for the induction of inflammatory mechanisms, the mAT1R Ab was administered to AT1R-knockout mice, deficient for both murine AT1Ra and AT1Rb. The mAT1R Ab-induced ear infiltration was reduced in the AT1R-deficient mice when compared with wild-type mice (figure 5D). In contrast, the isotype control IgG did not induce ear infiltration in the AT1R-deficient and wild-type mice. In terms of lung inflammation, the number of the available mice and the results obtained so far were too low or too few to yield significant differences between the AT1R-deficient and wild-type mice (data not shown).

Treatment with a monoclonal AT1R Ab induces skin and lung inflammation in vivo. Each mouse was injected intradermally into the ear repeatedly every other day from day 0 to day 12 (100 µg of anti-AT1R or isotype control IgG/injection). On day 14, the mice were sacrificed, and inflammation in the antibody-treated ear and in the lungs was evaluated histologically. (A) Representative histology of ears injected with mAT1R AB (right) or isotype IgG (IgG2a, left) (200×, scale bar=50 µm). Skin inflammatory scores of ears treated with mAT1R Ab or isotype IgG. The results are presented as mean±SD, and statistical analysis was performed using Student’s t-test (***p<0.001). (B) Representative micrographs of H&E-stained lung sections of mice treated with mAT1R AB (right) or isotype IgG (left). Incidence of pulmonary inflammation in mice injected with mAT1R Ab or isotype IgG (**p<0.01, Fisher exact test). (C) Characterisation of inflammatory infiltrations of ears and lungs of mice treated with mAT1R AB. T cells, B cells and neutrophils were detected by IHC. Representative pictures are shown. Scale bar=50 µm. (D) Representative micrographs of H&E-stained ear sections from wild-type and AT1R-deficient mice, which received mAb5.2a or murine IgG2a isotype control. Scale bar=100 µm. Quantified analysis of the severity of inflammation in the ear skin. Statistical analysis was performed using Student’s t test or Mann-Whitney U test. *p<0.05. AT1R, angiotensin receptor type 1.
Monoclonal AT1R Ab and murine IgG containing AT1R Abs specifically interact with AT1R-expressing cells from different species in vitro
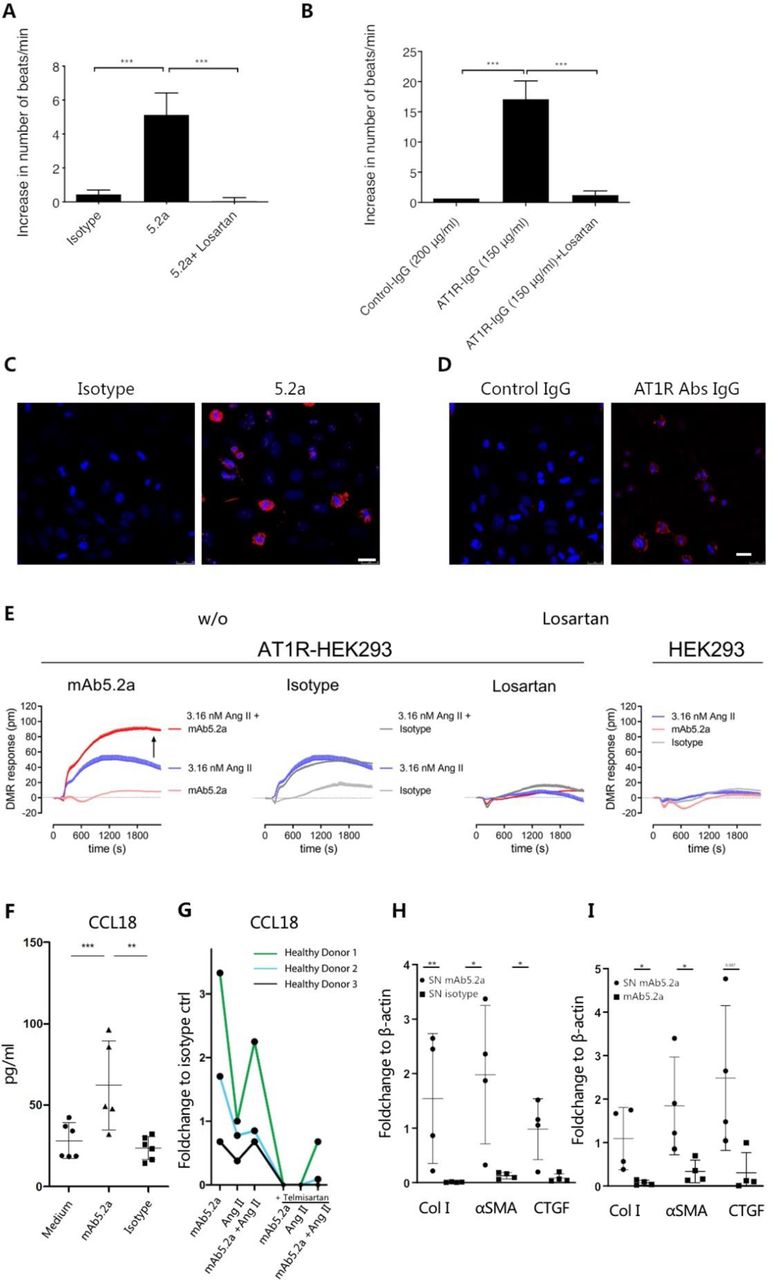
Four different cell types expressing AT1R were employed to examine interactions between AT1R and AT1R Abs in vitro.23 First, AT1R-expressing rat cardiomyocytes were treated with both mAT1R Ab as well as IgG derived from the AT1R-immunised mice as described before.24 While the mAT1R Ab increased the spontaneous cardiomyocyte beating frequency to on average 5 beats/minute compared with the isotype control, the stimulatory effect was completely abrogated by losartan, an AT1R antagonist (figure 6A, p<0.001). Similarly, IgG derived from AT1R-immunised mice increased the spontaneous cardiomyocyte beating frequency to on average 16 beats/minute compared with IgG from control mice (figure 6B). Again, this effect was completely inhibited by losartan (figure 6B, p<0.001), indicating specificity for AT1R. Further, following treatment of human epithelial (HEp-2) cells with the mAT1R Ab and IgG derived from AT1R-immunised mice, deposition of IgG onto cell membrane was observed in comparison to isotype control or IgG from control-immunised mice (figure 6C,D). To further validate the mAT1R Ab-induced effects, AT1R-transfected and naive human embryonic kidney (HEK293) cells were used in a label-free optical whole-cell biosensing assay based on detection of DMR.21 22 This technology platform captures morphological changes that occur in living cells as a consequence of ligand receptor interaction.21 22 Here, the mAT1R Ab as well as the isotype control did not show an effect when used alone (figure 6E). However, in contrast to the isotype control, the DMR response by Ang II was elevated in the presence of the mAT1R Ab. All DMR responses were completely abrogated by AT1R blockade through losartan and were undetectable in cells without exogenous enrichment of AT1R (figure 6E). The results indicate enhancement of Ang II effects by AT1R Abs in a strictly AT1R-dependent manner. Finally, in blood monocytes from healthy donors, the mAT1R Ab induced CCL18, but not the isotype control (figure 6F). The induction of CCL18 by the mAT1R Ab alone and in combination with Ang II was higher compared with Ang II alone. The effect was abrogated by the AT1R antagonist telmisartan (figure 6G). Conditioned supernatant of mAT1R Ab-stimulated monocytes induced higher expression of α-SMA, connective tissue growth factor (CTGF) and collagen I in primary dermal fibroblast compared with the isotype control (figure 6H, online supplemental figure 7) or compared with the stimulation by the mAT1R Ab alone (figure 6I, online supplemental figure 7), pointing towards a pivotal role of monocytes and the mAT1R Ab/AT1R axis for fibrosis. Taken together, the data indicate that in vitro AT1R Abs interact with AT1R expressed by rodent and human cells. Moreover, AT1R Abs activate AT1R or act in agonistic fashion together with Ang II. These interactions turn into altered functions such as increased heartbeat or elevated chemokine release.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
AT1R Abs interact with AT1R-expressing rodent and human cells in vitro. (A) Increase of the beating frequency of cardiomyocyte induced by the mAT1R ab or (B) by IgG isolated from AT1R-immunised mice and blockade by 1 µM losartan. The results are presented as mean±SD, statistical analyses were performed using one-way analysis of variance and Bonferroni’s multiple comparisons test (***p<0.001). (C) Uptake of an mAT1R Ab (clone 5.2a) or (D) of IgG isolated from AT1R immunised mice by human epithelial type 2 (HEp-2) cells (630×, scale bar=25 µm) detected by IF showing mouse IgG deposits in red and nuclei (DAPI) in blue. (E) Label-free biosensing of human AT1R-overexpressing HEK293 cells exposed to mAb5.2a or IgG2a isotype control (50 µg/mL) either alone or with agonist Ang II measured by DMR. Arrow in the left panel indicates enhancement of Ang II signal by mAb5.2a. AT1R specificity is shown by pretreatment with 10 µM AT1R antagonist losartan and by stimulating cells without exogenous AT1R enrichment showing no Ab response. Representative kinetic recordings are shown as mean+SEM of a triplicate determination. (F) Human peripheral monocytes were stimulated with recombinant AT1R monoclonal antibody 5.2 a, IgG2a isotype control and unconditioned medium control or (G) with recombinant AT1R mAb 5.2a, Ang II or both with or without telmisartan. CCL18 levels in supernatants (SN) were measured. (H, I) Densitometric analysis of Western blots show collagen 1, α-SMA and CTGF expression of monocyte SN-treated or mAT1R Ab-treated dermal FBS (n=4) relative to β-actin (image Studio Lite). For (H) conditioned SN from mAb 5.2a-stimulated or isotype-stimulated human blood monocytes or for (I) FB stimulated with conditioned SN from mAb 5.2a or with the mAb5.2a alone. Ratio t-test was used to test statistical significance. All data are shown as mean±SD.
Discussion
Interactions between AT1R Abs and AT1R expressed by tissue-resident and/or circulating immune cells are suspected to contribute to vascular, fibrotic and inflammatory processes, which are also key features in SSc.11 15 25 26 Here, immunisation of C57BL/6J mice with ME of human AT1R-overexpressing CHO cells induced high levels of AT1R Abs accompanied by lung and skin inflammation. Lymphocytic alveolitis, indicative of ILD, elevated Smad2/3 signalling and collagen expression, indicative of skin fibrosis, as well as mild endothelial apoptosis were detected. These manifestations were not observed in control mice receiving ME without AT1R overexpression. CD4+ T cells and B cells are indispensable for the AT1R-induced phenotype and immunisation with an AT1R peptide predicted to bind best to MHC class II also induced a phenotype. Moreover, local and repeated immunisation of C57BL/6J mice with an mAT1R Ab generated based on AT1R immunisation also induced skin and lung inflammation. Of note, when AT1Ra/b KO mice were immunised with the mAT1R Ab, skin inflammation was reduced when compared with wild type, suggesting interaction between AT1R Abs and AT1R. These data support former in vitro studies showing a correlation between AT1R ab levels and lymphocyte migration.17 The leucocyte infiltration observed in lung and skin following AT1R immunisation or local application of an mAT1R Ab for the first time implicates that anti-GPCR Abs contribute to immune cell trafficking in vivo. Therefore, our study supports the idea that AT1R Abs and possibly other anti-GPCR Abs play a role in the regulation of immune cell migration to specific organs.27
As mentioned above, following immunisation with AT1R, increased collagen expression and skin thickening were also observed. Interestingly, infusion of Ang II in the skin of C57BL/6J mice results in local inflammation and fibrosis.28 Taking into account that AT1R Abs have agonistic and synergistic effects to Ang II, such AT1R Abs could indeed augment the effect of Ang II to develop skin fibrosis.29 Accordingly, supernatants from mAT1R Ab-stimulated monocytes induced α-SMA, CTGF and collagen I in primary dermal fibroblasts.
Here, ME from CHO cells overexpressing AT1R were used for immunisation in order to mimic the natural structure of a transmembrane protein. Applying this approach, functionally active AT1R Abs were induced in vivo. However, in patients with SSc, high AT1R Ab levels were shown to be associated with vascular complications.11 15 Herein, although perivascular inflammation and endothelial apoptosis were observed, we did not detect signs of obliterative vasculopathy. In addition, the degree of skin fibrosis does not correlate with the Ab levels, which suggests additional mechanisms. Both obliterative vasculopathy and fibrosis are often associated with the presence of Th2 cytokines, which were shown to be predominant in SSc.30 31 These pathologies also require a bidirectional cross-talk between immune and stromal cells.32 When using complete/incomplete Freund’s adjuvant (CFA/IFA) like herein, a strong Th1 immune response will be induced.33 Therefore, we cannot exclude that a specific cytokine microenvironment or pathogenic factors inducing Th2 cytokines are required to develop obliterative vasculopathy or more robust fibrosis. In the future, the mouse model established herein could be employed to analyse potentially important further pathways such as the specific role of T cells, for example, by adoptive transfer experiments, or of specific cytokines. Accordingly, as suggested by first experiments on Th2 background, our immunisation strategy could pave the way to establish animal models, for example, for SSc with a more robust phenotype for fibrosis and vasculopathy. The results presented here fit to our novel humanised mouse model, where transfer of peripheral blood mononuclear cells (PBMC) derived from patients with SSc, but not from rituximab-treated patients with SSc, induced inflammatory lung disease indicating the role of B cells and Abs in the early inflammatory phase of SSc.34 Moreover, as shown in our recent study, AT1R Abs were among the best to identify severe COVID-19 infection, which indicates their relevance in lung inflammation also due to other diseases.35
In vitro studies using rat cardiomyocytes, HEp-2 cells and human monocytes yielded that the interaction with both the murine mAT1R Ab and the model-derived IgG containing polyclonal AT1R Abs induced cellular responses which can be blocked by AT1R antagonists.36 Moreover, the murine mAT1R Ab exhibited agonistic effects alone and in combination with Ang II depending on the cellular function as indicated by stimulation of cardiomyocytes, primary monocytes or by measuring morphological changes of the AT1R-transfected HEK293 cells via DMR.37 In the latter, induction of a signal by stimulation with both Ang II and mAT1R Abs, but not by mAT1R Ab alone, may fit to the idea that GPCR Abs act allosteric and modulate the effect of an orthosteric ligand such as Ang II towards a prolonged stimulation of the receptor.14 29 Besides, it has been reported that AT1R Abs from patients with preeclampsia limit the AT1R internalisation.38 Thus, future experiments could be envisioned to determine if AT1R Abs affect AT1R internalisation by various cells as well and if or how this influences G protein signalling. Overall, interactions between Ang II, AT1R Abs and AT1R need to be investigated in more detail. As recently demonstrated, AT1R Abs as well as other GPCR Abs feature disease-specific interactions with other proteins and receptors that are very likely to affect their specific signalling. Accordingly, a strong cross-talk of AT1R Abs to the endothelin receptor type-1 has been described.15 Additional work is necessary to find out whether those cross-talks can become a target of therapeutic intervention.27
Taken together, induction of AT1R Abs went along with signs of skin and lung inflammation, in particular lymphocytic alveolitis, perivascular infiltrations, endothelial apoptosis and skin fibrosis. Moreover, the induction of skin and lung inflammation following immunisation with the mAT1R Ab indicates that AT1R Abs can indeed directly contribute to the development of symptoms found in SSc. AT1R Abs stimulated AT1R across species. Thus, our mouse model offers a new perspective to examine the role of GPCR Abs in vivo alone and in combination with other ligands or factors. AT1R Abs should come into the focus to develop new therapies for diseases associated with high levels of AT1R Abs.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and for donation of fibroblasts, each individual donor submitted an informed written consent and the ethics committee of the Medical Faculty of the University of Lübeck approved the study (21-191). Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors would like to thank Prof. Thomas Walther, University College Cork, Ireland, and University Medicine Greifswald, Germany, for kindly providing AT1R-deficient mice.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
XY, JY and XW contributed equally.
Correction notice This article has been corrected since it published Online First. The affiliations and equal contributor statement have both been updated and Prof Petersen has been added as corresponding author. Also, the open access licence has been updated to CC BY. 25th May 2023.
Contributors GR, XYu and FP were involved in the conception and design of the study. XYue, XW, JY, BK, IS, GW, AK-S, JEH, JZ, LMD, NM, EK, AMH, XD, LW, LZ, GH and AM performed the experimental work, collected and analysed the data. HH, KS-F and JJ contributed to developing and providing essential materials (ME and ELISA). XYu, FP, GR and XYue were involved in drafting the manuscript and all authors were involved in revising the manuscript. GR and FP ae the guarantors.
Funding This work was supported by funding from the German Research Foundation (DFG: RTG 1727, RTG 2633, RI 1056 11-1/2, Excellence Cluster 'Precision Medicine in Inflammation', TI4), the German Ministry of Education and Research (BMBF, Mesinflame consortium, project no 5, funding grant no 01EC1901D), the Eppenauer Gutzeit Foundation, the Edith-Busch Foundation and the German Centre for Lung Research (DZL).
Competing interests HH is the owner and GR is an advisor of the company CellTrend GmbH, which produced the ME and the tests for the detection of AT1R Abs. IS and GW are shareholders of Berlin Cures GmbH, where the rat cardiomyocyte assay was performed.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.