Article Text

Abstract
Objectives To find causal genes for rheumatoid arthritis (RA) and its seropositive (RF and/or ACPA positive) and seronegative subsets.
Methods We performed a genome-wide association study (GWAS) of 31 313 RA cases (68% seropositive) and ~1 million controls from Northwestern Europe. We searched for causal genes outside the HLA-locus through effect on coding, mRNA expression in several tissues and/or levels of plasma proteins (SomaScan) and did network analysis (Qiagen).
Results We found 25 sequence variants for RA overall, 33 for seropositive and 2 for seronegative RA, altogether 37 sequence variants at 34 non-HLA loci, of which 15 are novel. Genomic, transcriptomic and proteomic analysis of these yielded 25 causal genes in seropositive RA and additional two overall. Most encode proteins in the network of interferon-alpha/beta and IL-12/23 that signal through the JAK/STAT-pathway. Highlighting those with largest effect on seropositive RA, a rare missense variant in STAT4 (rs140675301-A) that is independent of reported non-coding STAT4-variants, increases the risk of seropositive RA 2.27-fold (p=2.1×10−9), more than the rs2476601-A missense variant in PTPN22 (OR=1.59, p=1.3×10−160). STAT4 rs140675301-A replaces hydrophilic glutamic acid with hydrophobic valine (Glu128Val) in a conserved, surface-exposed loop. A stop-mutation (rs76428106-C) in FLT3 increases seropositive RA risk (OR=1.35, p=6.6×10−11). Independent missense variants in TYK2 (rs34536443-C, rs12720356-C, rs35018800-A, latter two novel) associate with decreased risk of seropositive RA (ORs=0.63–0.87, p=10−9–10−27) and decreased plasma levels of interferon-alpha/beta receptor 1 that signals through TYK2/JAK1/STAT4.
Conclusion Sequence variants pointing to causal genes in the JAK/STAT pathway have largest effect on seropositive RA, while associations with seronegative RA remain scarce.
- rheumatoid arthritis
- autoantibodies
- polymorphism, genetic
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. The GWAS summary statistics are available at https://www.decode.com/summarydata/. Sequence variants passing GATK filters will be deposited in the European Variation Archive (https://www.ebi.ac.uk/ena/data/view/). We used publicly available software (URLs listed further) in conjunction with the algorithms in the sequencing processing pipeline (whole-genome sequencing, association testing, RNA-sequence mapping and analysis, see methods description in Supplementary Information 2): BWA 0.7.10 mem (https://github.com/lh3/bwa); GenomeAnalysisTKLite 2.3.9 (https://github.com/broadgsa/gatk/); Picard tools 1.117 (https://broadinstitute.github.io/picard/); SAMtools 1.3 (http://samtools.github.io/); Bedtools v2.25.0-76-g5e7c696z (https://github.com/arq5x/bedtools2/); Variant Effect Predictor (https://github.com/Ensembl/ensembl-vep); Read_haps (http://github.com/DecodeGenetics/read_haps); In-silico prediction of missense variants (https://sites.google.com/site/jpopgen/dbNSFP).
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Although many genetic risk loci have been identified in rheumatoid arthritis (RA) overall, there are limited data available on the seropositive and seronegative subsets. Furthermore, most reported RA associations outside the HLA-locus are with common non-coding variants with low risk,which lack a compelling candidate gene mediating the effect on RA.
Key messages
What does this study add?
In this largest genome-wide association study on RA to date, we studied both RA overall and the seropositive and seronegative RA subsets and found several unreported sequence variants with large effect on the risk of seropositive RA, while associations with seronegative RA were scarce. Through a genomic, transcriptomic and proteomic analysis, we identified candidate causal genes for most signals and show that the majority of those associated with seropositive RA are in the interferon alpha/beta and IL-12/23 signalling networks. Furthermore, most sequence variants that confer the largest risk of seropositive RA point to causal genes encoding proteins in the JAK/STAT-pathway and have not been reported in RA before. This includes a missense variant in the STAT4 gene that confers 2.27-fold risk, larger than the lead signals at the well-known HLA-DRB1 and PTPN22 loci, and two unreported missense variants in the TYK2 gene, affecting levels of the interferon-alpha/beta receptor 1 (IFNAR1).
How might this impact on clinical practice or future developments?
These findings highlight how a multiomics approach can reveal causal genes. Our findings support treatment of seropositive RA with the already registered JAK and IL-6R inhibitors as well as CTLA4-Ig but also open for repurposing of other drugs that target proteins in the JAK/STAT-pathway, including inhibitors of FLT3, TYK2 and IFNAR1.
Introduction
Rheumatoid arthritis (RA) is a heterogeneous clinical syndrome that affects around 0.5%–1% of the general population. It is characterised by inflammatory polyarthritis and progressive joint damage if insufficiently treated.1 RA is divided into seropositive and seronegative RA, where around two-thirds of RA patients are in the seropositive subset, based on autoantibodies (rheumatoid factor (RF) and/or antibodies against citrullinated peptide antigens (ACPA)).1 2 Although many risk loci have been identified in previous genome-wide association studies (GWAS), most reported RA associations are with common non-coding variants that confer low risk and lack a compelling candidate gene mediating the effect on RA.1 3–6 The main exceptions are the shared epitope encoded by certain alleles of HLA-DRB1 and two missense variants in the PTPN22 (rs2476601-A) and TYK2 (rs34536443-C) genes.1 3
Previous GWAS have focused on RA overall,3–6 except for one study on ACPA-positive (n=1147) and ACPA-negative (n=774) RA that confirmed the strong association of HLA-DRB1 alleles with ACPA-positive RA but did not identify any genome-wide significant signals outside the HLA-locus7 and another report on ACPA-negative RA only (n=1922) that identified two genome-wide significant signals.8
Here, we searched for sequence variants outside the HLA-locus affecting the risk of RA overall, the seropositive and/or seronegative subsets of RA, using the largest GWAS study population to date in RA (31 313 cases and ~1 million controls) from six countries in Northwestern Europe and searched for candidate causal genes through a genomic, transcriptomic and proteomic analysis.
Methods
Study populations
Cases with RA were diagnosed by rheumatologists and/or captured through the nationwide Scandinavian rheumatology quality registries and/or the 10th revision of the International Statistical Classification of Diseases (ICD-10) code-based registration of all inpatient and outpatient healthcare visits (see four-digit based ICD-10 codes in table 1). If available, RF and anti-CCP measurement were used to define the seropositive/seronegative RA subsets, according to classification criteria.2 9
RA study populations from six Northwestern European countries included in the present study*
An overview of the study populations is provided in table 1. In the study populations from Iceland (3613 cases and 341 788 controls), UK Biobank (5798 cases and 402 767 controls of self-reported white British ancestry, confirmed by genetic analysis)10 and FinnGen (https://www.finngen.fi/en/access_results version R4: 4701 cases and 125 923 controls), RA cases were compared with the remaining non-RA individuals, with the Icelandic study covering a large part of the Icelandic population and the latter two being nationwide genetic cohort studies. From Sweden, we included: (1) the population-based EIRA case–control study (www.eirasweden.se) with 3436 newly diagnosed cases and 3058 controls matched for age, sex and geographical area from mid and Southern parts of Sweden. In addition, we included 7488 controls from the parallel Swedish EIMS study (ki.se/imm/eims-epidemiologisk-undersokning-av-riskfaktorer-for-multipel-skleros); (2) the RA cohort from Umea (n=1935) and 1156 controls from Umea biobank, matched for age and sex (www.umu.se/en/biobank-research-unit); and (3) the Swedish Rheumatology Quality Register Biobank (n=3287, www.srq.nu).
From Denmark, RA cases were identified in four study populations: (1) Danish Biomarker Protocol11 (n=2544 with samples in the Danish Rheumatological Biobank and clinical data in the Danish Rheumatology Quality Register, DANBIO)12 (2) the Copenhagen Hospital Biobank (n=3282), (3) the TARCID cohort (n=1826) and (4) the nationwide Danish Blood Donor Study (DBDS; 10 RA cases).13 Controls for these 7662 cases were age-matched and sex-matched non-RA individuals from DBDS (n=86 964).
From Norway, 881 RA cases from the Oslo RA cohort and 28 517 population-based controls from the Norwegian Mother, Father and Child Cohort Study were included.14 15
Patients were involved in the design and conduct of several of the studies that are included in this report.
Genotyping and multiomics analyses
For a detailed methodological description, see online supplemental information 2. In short, genotyping of all cohorts except UK Biobank and FinnGen was performed at deCODE genetics using the Illumina technology, and the sequence variants for imputation were identified through whole-genome sequencing of 67 645 individuals.
Supplemental material
We used logistic regression to test the association of ~64 million sequence variants with RA overall, the seropositive and the seronegative subset.16 Sequence variants were split into five classes based on their genome annotation, and the significance threshold for each class was based on the number of variants in that class,17 thereby adjusting for all ~64 million variants tested, maintaining an unadjusted significance threshold of 8×10−10. The primary signal at each genomic locus has the lowest Bonferroni-adjusted p value. Conditional analysis was used to search for possible secondary signals (<500 kB from the primary signal, excluding HLA-locus). We tested whether primary and secondary signals were in strong linkage disequilibrium (R2 >0.8) with top cis-eQTL variants for genes expressed in various tissues (online supplemental tables 5 and 6), and/or with levels of 4789 proteins in plasma (pQTL, SomaScan, Somalogic) in 35 559 Icelanders (online supplemental table 7).18–21
Supplemental material
Supplemental material
Supplemental material
We used the Ingenuity Pathway Analysis software (QIAGEN Inc) to evaluate whether there is experimental evidence for direct or indirect interaction between the proteins coded by candidate causal genes, supporting biological connection.
Results
Genome-wide association study
Of the 31 313 RA cases, 26 534 (84.7%) had information on serological status. Of these, 18 019 (67.9%) were seropositive and 8515 (32.1%) seronegative (table 1).
In separate meta-analyses of RA overall and the seropositive and seronegative RA subsets, we found in total 37 sequence variants at 34 non-HLA loci (online supplemental figure 1a–c), as summarised in table 2. Thus, we identified 25 lead signals for RA overall (online supplemental table 2), 33 for seropositive and 2 for seronegative RA (online supplemental table 3). When we searched for novel sequence variants, we adjusted for 82 independent sequence variants previously reported to associate with RA (p<5×10−8 in the largest meta-analysis to date),4 6 and 15 of the 37 sequence variants are previously unreported. The 15 novel associations are at 12 loci and six of those loci are previously unreported. Little heterogeneity was observed between the study populations (see online supplemental tables 2 and 3 (Phet ) and online supplemental figure 4 (average effect)).
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Sequence variants outside the HLA locus that associate with RA overall, seropositive (rheumatoid factor and/or anti-CCP antibody positive) and/or seronegative RA in GWAS meta-analysis within six Northwestern-European countries (table 1). Association results are shown for the lead signals for all three RA groups, and the heterogeneity between the seropositive and seronegative subsets.† Effect alleles with novel associations are marked with.*
Supplemental material
Replication of previously reported signals
We replicated 53 of the 82 previously reported variants (online supplemental table 1, correcting for multiple testing, p value threshold=0.05/82 variants /3 phenotypes=2.03×10−4). However, only 36 of the 82 variants were previously reported to be genome-wide significant in Europeans,4 6 and we replicated 34 of these 36 variants (94%).
Comparison of RA subsets
The heritability estimates (total observed scale h2) were higher for seropositive RA (0.19 (0.022)) than for seronegative RA (0.099 (0.019)). For a substantial proportion of the RA-associated sequence variants, their effect was greater on seropositive RA than seronegative RA risk (table 2, figure 1). However, the genetic correlation between seropositive and seronegative RA was high (rg 0.87, SE 0.13, p=4.5×10−12 (online supplemental table 9).
Supplemental material

Effects of the lead sequence variants associated with seropositive RA (18 019 cases) compared with RA overall (31 313 cases, left graph) and seronegative RA (8515 cases, right graph). The x-axis and the y-axis show the logarithmic estimated ORs for the associations with the three phenotypes. All effects are shown for the RA risk increasing allele based on current meta-analysis of study population from six countries in Northwestern Europe (table 1). Error bars represent 95% CIs. The red line represents slope (SD) based on a simple linear regression through the origin using MAF (1-MAF) as weights. See further results in table 2 and online supplemental tables 2; 3.
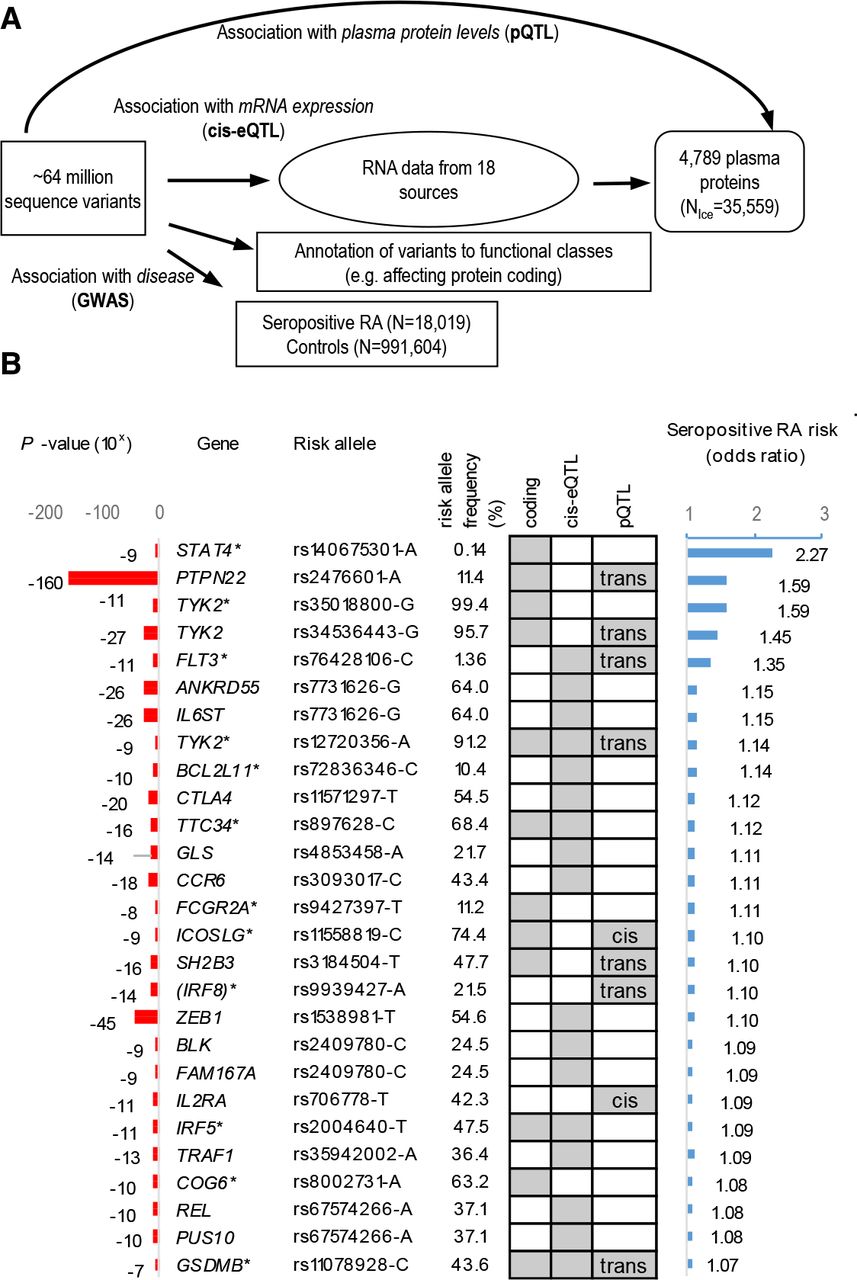
Identification of sequence variants that associate with seropositive RA and the multiomics approaches used to recognise candidate causal genes. (A) schematic overview of the experimental approach used to identify sequence variants that associate with seropositive RA and their systematic annotation, applying multiomics approach to identify candidate causal genes, that is, based on whether lead variants or correlated variants (R2 >0.8) affect protein coding (online supplemental tables 2–4), mRNA expression (cis-eQTL (online supplemental tables 5 and 6)) or levels of proteins in plasma (pQTL (online supplemental table 7)). (B) Out of 33 lead variant associations outside the HLA-locus (online supplemental table 3), 25 candidate causal genes were identified as listed, ranked by effect (OR). All effects are shown for the risk increasing allele based on GWAS in RA study populations from Northwestern Europe (table 1). Associations that are previously unreported in RA are marked with *. Grey boxes highlight where data point to a candidate causal gene. GWAS, genome-wide association study; RA, rheumatoid arthritis.
Supplemental material
Genomic, transcriptomic and proteomic analysis of lead signals
We searched for candidate causal genes with an omics approach (figure 2A) and evaluated the effect of lead signals (or correlated variants, R2 >0.8) on amino acid sequence (online supplemental tables 2–4), mRNA expression (cis-eQTL (online supplemental tables 5 and 6) and/or plasma levels of proteins (pQTL (online supplemental table 7). This yielded a total of 27 candidate causal genes in RA overall and/or its subsets.
Seropositive RA
Twenty-four of the 33 lead signals in seropositive RA pointed to 25 candidate causal genes, as shown in figure 2B ranked by effect. The one with the largest effect is a rare (MAF=0.14%) missense variant in the STAT4 gene (rs140675301-A, Glu128Val) that associates with 2.27-fold increased risk (p=2.1×10−9, table 2 and figure 2B). Rs140675301-A is the first coding variant identified at the STAT4 locus that associates with RA and has not been reported in any disease before. This signal is independent (online supplemental table 8) of the common lead STAT4 intronic variant (rs4853458-A), which is strongly correlated (R2=1) with other intronic variants in STAT4, previously reported to associate with RA22 23 (figure 3A and online supplemental table 1). STAT4 contains six domains that have different functions, and the rare missense rs140675301-A variant leads to an amino acid change from negatively charged, hydrophilic, glutamic acid to non-polar hydrophobic valine at position 128 (Glu128Val) in a loop on the surface of the protein (figure 3B), between the N-terminal domain and the helical coiled coil domain. The coiled coil domain provides a carbonised hydrophilic surface that binds to regulatory factors.24 The amino acid sequence and secondary structure of the loop is highly conserved between species (figure 3C) and within the family of STAT proteins,24 25 indicating its importance for the function of STAT4. Tetramer formation of STAT at DNA binding sites is necessary for full transcriptional activation of many of its target genes,26 and STAT without the N-terminal domain cannot form tetramers.27
Supplemental material
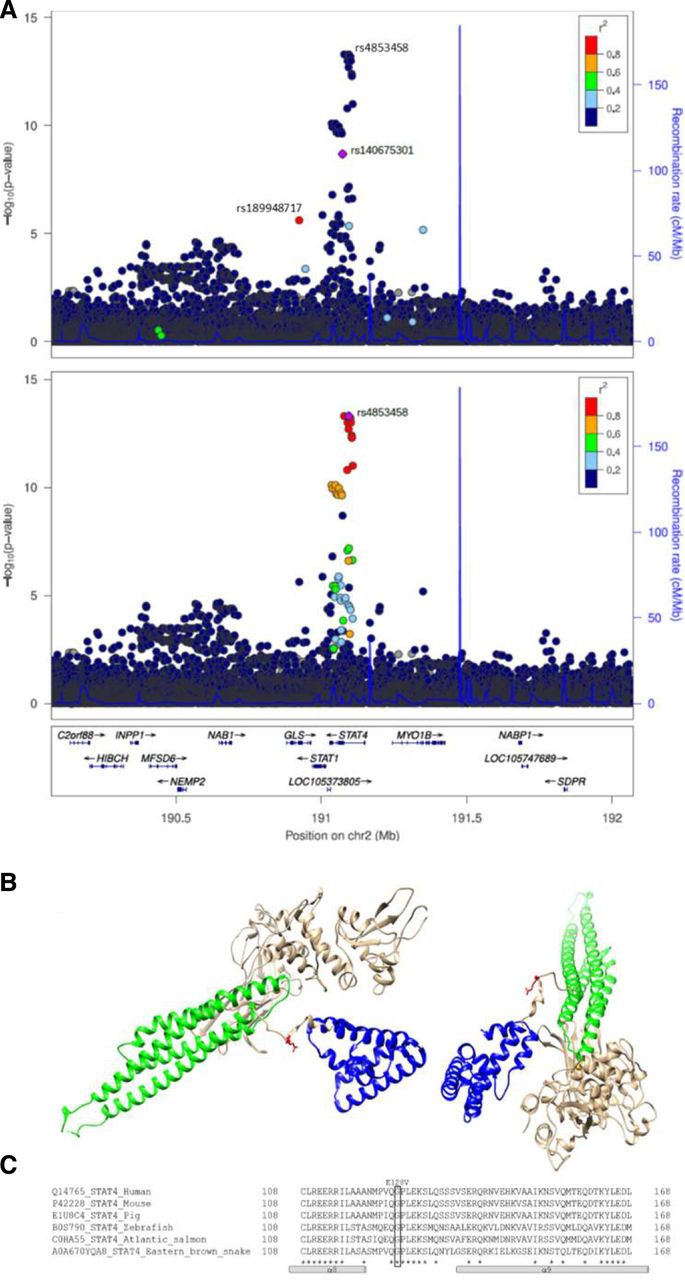
STAT4 missense variant rs140675301 is associated with seropositive RA (18 019 cases), is not correlated with previously reported variants at the locus and leads to an amino acid change in a highly conserved area of the protein. (A) Locus plot for the association of variants at the STAT4 locus with seropositive RA. The upper graph illustrates that the intronic variant rs4853458, that is the lead variant at the locus, is not correlated (r2 <0.2) with the missense variant rs140675301, that is coloured in purple. The missense variant rs140675301 is only highly correlated (r2 >0.8) with one variant, the intronic variant rs189948717 (coloured in red), that has less effect (seropositive RA: OR=1.81, p=3.69×10−6). Neither of these variants have previously been reported in any disease. The lower graph highlights that the lead variant at the locus (rs4853458, coloured in purple) has many correlated variants, coloured by degree of correlation (r2) with rs4853458. (B) Secondary structure of STAT4 (viewed from two angles) based on a structural model with STAT1 crystal structure (PDB code: 1yvl.1.A (Mao et al, Molecular Cell 2005;17:761–71) as template. Glu128Val (red) is located in a loop connecting the N-terminal domain (blue), important for tetramer formation of STATs and nuclear translocation, and the coiled coil domain (green), which provides a carbonised hydrophilic surface that binds to regulatory factors.24 α-Helices are drawn as cylinders. Invariant residues are marked with asterix. (C) multiple sequence alignment of the conserved STAT4 loop between the N-terminal domain (α8) and the coiled coil (α9) domain, performed with Clustal omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). RA, rheumatoid arthritis.
The second largest effect on the risk of seropositive RA had the well-known missense variant rs2476601-A in the PTPN22 gene, followed by a novel missense variant in the TYK2 gene (rs35018800-A, Ala928Val), encoding tyrosine kinase 2, which is a member of the JAK/STAT-pathway like STAT4. This rare (MAF=0.60%) missense variant in TYK2 conferred reduced risk of seropositive RA (OR=0.63, p=1.4×10−11), independently of a known missense variant in TYK2 (rs34536443-C, Pro1104Ala, MAF 4.3%), which we also found to decrease the risk of RA overall (OR=0.75, p=2.5×10−29), and here, we extend this association to the seropositive RA subset (OR=0.69, p=2.7×10−27; table 2, online supplemental table 3 and online supplemental figure 2). In addition, we identified a common missense variant in TYK2 that independently associated with reduced risk of seropositive RA (rs12720356-C, Ile684Ser, MAF=8.82%, OR=0.87, p=2.3×10−9). Analysis of the plasma proteome (online supplemental table 7) showed that the minor alleles of the variants encoding both Ile684Ser and Pro1104Ala in TYK2 are the only sequence variants that associate in trans with plasma levels of interferon alpha/beta receptor 1 (IFNAR1, Ile684Ser: effect=−0.19 SD, p=7×10−25; Pro1104Ala, effect=−0.13 SD, p=6×10−10). These variants did not associate with levels of any other plasma protein measured. Notably, both the missense variants in TYK2 and STAT4 are predicted to damage the function of the encoded protein (online supplemental table 4).
Supplemental material
An intronic variant (rs76428106-C) in the FLT3 gene, encoding another tyrosine kinase receptor that signals through the JAK/STAT-pathway, conferred 35% increase in risk of seropositive RA (p=6.6×10−11). This is in accordance with our previous report, where we discovered this variant in a GWAS on autoimmune thyroid disease and found that it also associated nominally with the risk of seropositive RA (OR=1.41, p=4.3×10−4) and with increased levels of 22 proteins in plasma (trans-pQTL), including the FLT3 ligand18 (online supplemental table 7). rs76428106-C associated with increased mRNA expression of FLT3 in lung tissue (beta=0.82 SD, p=1.3×10−10, online supplemental table 6).
We performed a network analysis of the 25 seropositive RA candidate causal genes and found that 18 of them encode proteins that are linked in the same network (online supplemental figure 3), either through direct protein–protein interaction (eg, STAT4-TYK2, PTPN22-IRF5 and FLT3-SH2B3) or indirectly (eg, one affecting the level of another). Other molecules that are central in this network, and directly interact with proteins encoded by the candidate genes, are interferon alpha/beta and IL12/IL-23.
Supplemental material
Among the other candidate causal genes, we also identified novel loss-of-function variants in genes encoding molecules in this network, although with more modest effect on seropositive RA risk (table 2 and figure 2B). This includes a splice-donor variant in the IRF5 gene (rs2004640-G, OR=0.92, p=1.44×10−11) that encodes interferon regulatory factor 5. IRF5 rs2004640-G association with decreased risk of seropositive RA was independent from previously reported non-coding variants at the IRF5 locus (online supplemental table 1) and rs2004640-G is also associated with decreased mRNA expression of IRF5 in several tissues (online supplemental table 6). Other novel coding variants pointing to putative causal genes were missense variants in ICOSLG (rs11558819-T, OR=0.91, p=1.56×10−9) encoding ICOS ligand and TTC34 (rs897628-T, OR=0.90, p=3.28×10−16). TTC34 encodes tetratricopeptide repeat protein 34 that has an unknown role in the pathogenesis of RA and belongs to another network that includes the remaining seven candidate causal genes for seropositive RA (online supplemental figure 3).
Seronegative RA
Both signals in seronegative RA were also found in seropositive RA and pointed to causal genes: a missense variant rs2476601-A in PTPN22 and intronic variant rs7731626-A in ANKRD55 (table 2 and online supplemental tables 2; 3). PTPN22 rs2476601-A associated with plasma levels of several proteins (trans-pQTL), and it was the only variant in the genome to affect the levels of these proteins (online supplemental table 7). ANKRD55 rs7731626-A associated with a decreased risk of RA and its subsets and a decreased mRNA expression in whole blood of two neighbouring genes at the locus: ANKRD55 and IL6ST.
RA overall
The lead signals pointing to causal genes in RA overall were also identified in the seropositive subset (table 2), with two exceptions: missense variants in DNASE1L3 (rs35677470-A) and RIN3 (rs117068593-T) (online supplemental table 2). Both these missense variants are predicted to damage the function of the encoded protein (online supplemental table 4). DNASE1L3 rs35677470-A is a known signal in RA, but the RIN3 locus has to our knowledge not been reported to associate with any disease before. It encodes Ras and Rab interactor 3 that functions as a guanine nucleotide exchange factor of unknown relevance in RA.
Discussion
In this largest GWAS study on RA to date, we studied both RA overall and the seropositive and seronegative RA subsets and found 37 sequence variants of which 15 were previously unreported. Several of these have large effect on seropositive RA risk, while only two signals were identified in the seronegative subset, both previously reported in RA overall. Through a multiomics approach, we identified candidate causal genes for most signals and show that the majority of those associated with seropositive RA are in the interferon alpha/beta and IL-12/23 signalling networks, with largest risk associated with sequence variants in genes encoding proteins in the JAK/STAT pathway.
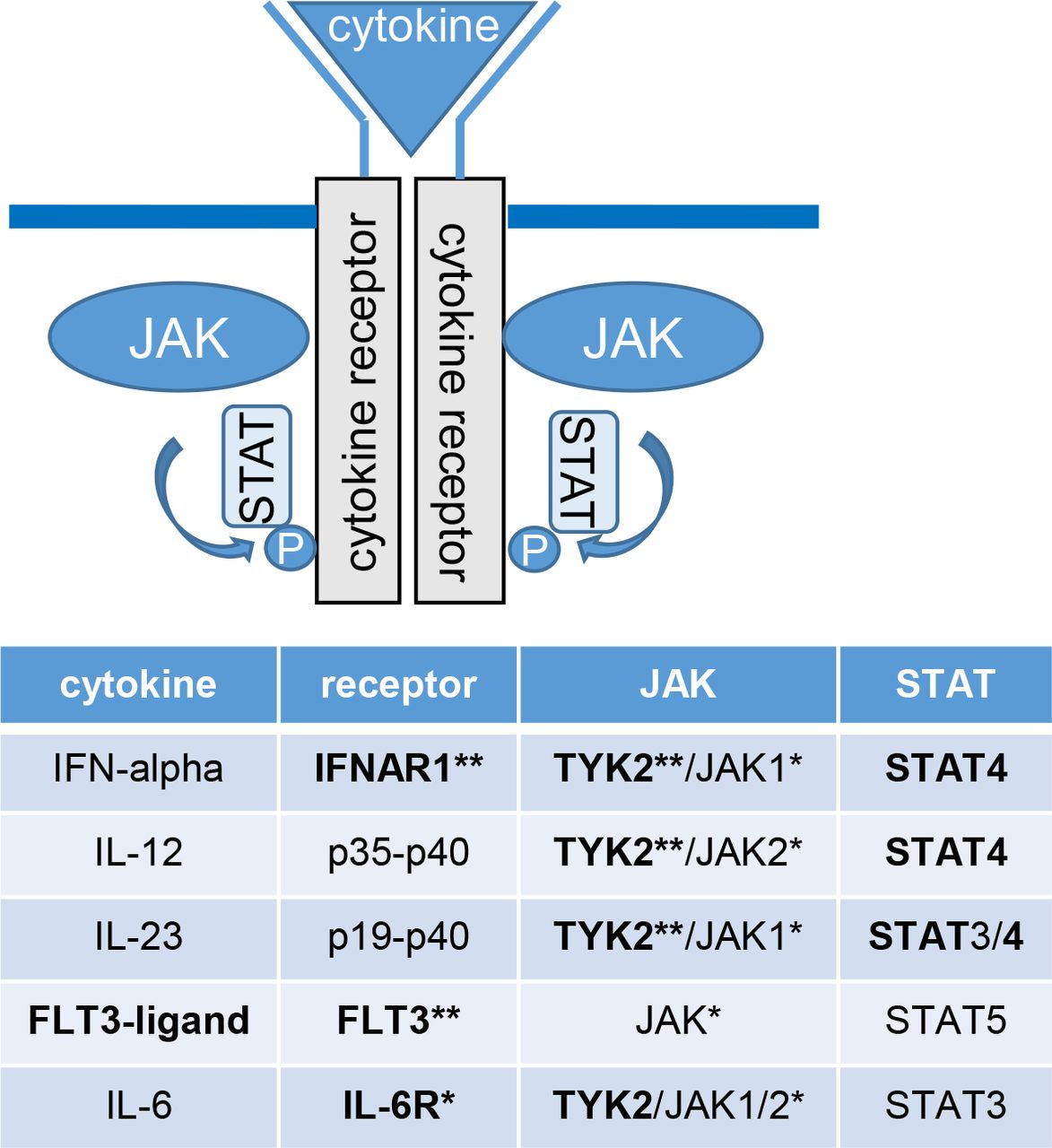
Novel missense variant in the STAT4 gene (rs140675301-A) confers 2.27-fold increased risk that is higher risk than any previously reported RA association, including the well-known HLA-DRB1 shared epitope and the lead missense variant at the PTPN22 locus. Although the STAT4 locus has been reported in genome-wide studies, this is the first STAT4 coding variant found to associate with RA. This coding variant points directly to STAT4 as the causal gene at the locus. It has not been reported for any other disease before, and we found that it leads to an amino acid change in a surface loop of the protein that is highly conserved, thereby underscoring its importance for STAT4 function. STAT4 encodes STAT4, a cytoplasmic transcription factor that regulates gene expression through the JAK/STAT-pathway.28 It is phosphorylated in response to various cytokines and displacement of the N-terminal and coiled coil domains within the protein structure could interfere with DNA binding, transcriptional activation and/or target selectivity. As highlighted in the network analysis and illustrated in figure 4, both interferon alpha, IL-12 and IL-23, signal through STAT4 via TYK2/JAK1 and TYK2/JAK2.29 Another RA-associated variant in STAT4 (rs7574865-T, R2=0.99 to lead intron variant rs4853458-A)23 increases IL-12-induced IFN-γ production in T cells.30 STAT4 is expressed at inflammatory sites in activated peripheral blood monocytes, fibroblasts, dendritic cells and macrophages and also in synovial macrophages and dendritic cells from patients with seropositive RA.28 31–34 Furthermore, reduced expression of STAT4 has been observed in RA patients that have responded well to disease-modifying treatment.32 Thus, STAT4 may have a central role in the inflammatory cascade in joints of RA patients.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The JAK-STAT pathway. The figure and table shows which receptors, JAK and STAT subtypes certain cytokines bind to, highlighting proteins encoded by and/or affected by causal genes in seropositive RA, based on the multiomics analysis of sequence variants associated with risk of seropositive RA (shown in bold). Binding of a cytokine to its receptor activates the associated Janus kinases (JAK). The JAK in turn phosphorylates (P) the receptor, which provides a docking for signal transducers and activators of transcription (STATs) and other signalling molecules to bind to the receptor. STATs also become phosphorylated and translocate to the nucleus, where they regulate gene expression. *Protein targeted by drugs that are registered for RA. **Proteins targeted by drugs registered or in pipeline for other diseases. RA, rheumatoid arthritis.
Tyrosine kinase 2, encoded by the TYK2 gene, is another key molecule in the JAK/STAT pathway that regulates signal transduction pathways downstream of the receptors for several cytokines, including interferon alpha/beta and IL-23/IL12 as described previously. We found that three independent coding variants in TYK2 associated with 25%–37% reduced risk of seropositive RA, and they associated with lower plasma levels of the IFNAR1 receptor for interferon-alpha/beta. Accordingly, one of the missense variants (Pro1104Ala) is located in the catalytic kinase domain of TYK2 and has previously been shown to reduce signalling through IFNAR1.35
TYK2 also mediates the signalling of IL-6, IL-10 and IL-4/IL-13.36 IL-6 signals through the IL-6 receptor (IL-6R), thereby inducing IL6ST homodimerisation and activation of TYK2/JAK1/2 and STAT3 signalling pathway (figure 4), known to play a role in RA.37 The intronic variant rs7731626-A in ANKRD55 associated with a reduced risk of both seropositive and seronegative RA and also reduced expression of ANKRD55 and IL6ST. The effect on IL6ST expression and its biological function points to IL6ST as a candidate causal gene at that locus. Accordingly, drugs inhibiting IL-6R are effective in RA.38
The FLT3 receptor is another activator of the JAK/STAT pathway that signals through STAT539 (figure 4), and an intronic variant in the FLT3 gene (rs76428106-C) conferred 35% increase in risk of seropositive RA. This confirms a non-genome-wide significant signal in our previous report, in which we identified this variant as a strong risk factor for autoimmune thyroid disease and found that it generates a cryptic splice site, introducing a stop codon in 30% of transcripts that are predicted to encode a truncated protein, lacking its tyrosine kinase domains.18 FLT3 encodes fms-related tyrosine kinase 3 receptor, a key regulator in the development of monocytes and dendritic cells. The cell-surface receptor is expressed on common dendritic cells and lymphoid/myeloid progenitors that give rise to both classical and plasmacytoid dendritic cells, which produce large amount of interferons when activated.40 As previously reported, FLT3 rs76428106-C increases plasma levels of the FTL3 ligand,18 and RA patients have increased levels of FLT3 ligand both in serum and synovial fluid of inflamed joints.41 42 FLT3 ligand deficient mice are protected against collagen-induced arthritis,42 and in a mouse model of collagen-induced arthritis, an oral inhibitor of FLT3/JAK2/c-Fms was found to block signalling through TYK2 and STAT4 and decrease both inflammation and bone resorption.43
Yet another variant affecting interferon signalling is a splice-donor variant in the IRF5 (rs2004640-G) gene that encodes interferon regulatory factor 5 and reduced both RA risk and IRF5 expression. IRF5-rs2004640-G has not been reported in GWAS on RA before, although the locus is known, and a tentative association was reported in a meta-analysis of candidate gene studies (4818 cases, p=0.003).44
The size and homogeneous background of the study populations, with ~64 million sequence variants derived from over 67 thousand whole-genome sequenced individuals, increases the likelihood to detect rare and low-frequency sequence variants that associate with disease. Furthermore, we were able to test their functional relevance through analysis of RNA sequence and plasma proteome. However, it remains to be seen whether the sequence variants associate with RA in populations of another ancestries.
The SNP-based heritability estimate for seropositive RA was the same as in a previous study (0.19),45 while lower for seronegative RA (0.099) where previous findings are scarce.46
In addition to the causal genes highlighted previously, the network analysis illustrated how majority of all candidate causal genes encode proteins in the interferon alpha/beta and IL-12/IL-23 signalling network. Furthermore, we observed a consistent direction of the effect on seropositive RA risk, gene expression and protein levels in plasma, indicating that increased signalling through the JAK/STAT-pathway is central in the inflammatory cascade in seropositive RA. Our findings are in line with the documented effectiveness of IL-6 receptor and JAK inhibitors (baricitinib, tofacitinib, filgotinib and upadacitinib) as well as CTLA4-Ig in RA.1 36 38 47 Furthermore, there are inhibitors of other proteins in this pathway that are in development or already marketed for other diseases but have to our knowledge not been tested for treatment of RA, including FLT3 inhibitors used to treat acute myeloid leukaemia and other cancer forms,48 TYK2 inhibitors that show promising results in clinical trials for psoriatic arthritis49 and IFNAR1 inhibitors in systemic lupus erythematosus.50
In summary, through a large genome, transcriptome and proteome analysis of RA and its subsets, we identified new RA risk loci and highlight candidate causal genes at the majority of RA-associated loci. Most sequence variants have larger effect on the risk of seropositive than seronegative RA. Majority of those with largest effect on RA risk have not been reported before and point to candidate causal genes encoding proteins in the network of interferon alpha/beta and IL-12/IL-23 that signal through the JAK/STAT pathway. Together, these data thus shed light on the molecular mechanism affected by most non-HLA sequence variants that predispose to seropositive RA. In contrast, the genetic background of seronegative RA remains largely unexplained.
Supplemental material
Supplemental material
Data availability statement
Data are available in a public, open access repository. All data relevant to the study are included in the article or uploaded as supplementary information. The GWAS summary statistics are available at https://www.decode.com/summarydata/. Sequence variants passing GATK filters will be deposited in the European Variation Archive (https://www.ebi.ac.uk/ena/data/view/). We used publicly available software (URLs listed further) in conjunction with the algorithms in the sequencing processing pipeline (whole-genome sequencing, association testing, RNA-sequence mapping and analysis, see methods description in Supplementary Information 2): BWA 0.7.10 mem (https://github.com/lh3/bwa); GenomeAnalysisTKLite 2.3.9 (https://github.com/broadgsa/gatk/); Picard tools 1.117 (https://broadinstitute.github.io/picard/); SAMtools 1.3 (http://samtools.github.io/); Bedtools v2.25.0-76-g5e7c696z (https://github.com/arq5x/bedtools2/); Variant Effect Predictor (https://github.com/Ensembl/ensembl-vep); Read_haps (http://github.com/DecodeGenetics/read_haps); In-silico prediction of missense variants (https://sites.google.com/site/jpopgen/dbNSFP).
Ethics statements
Patient consent for publication
Ethics approval
This research has been conducted using the UK Biobank Resource (application licence number 24898, REC Reference Number: 06/MRE08/65), and the study was approved by the National Bioethics Committees in Iceland (approval no. VSN-15-045 and VSN-16-042), Sweden (approval no. 96-174, 2006/476-31/4, 2007/889-31/2, 2012/2070-31/2, 2015.1746-31.4 and 04-252/1-4), Denmark (Danish Data Protection Agency (general approval number 2012-58-0004 and local number: RH-2007-30-4129/ I-suite 00678) and the National Committee on Health Research Ethics (NVK-1700407, NVK-1803863 and H-2-2014-086)) and Norway (Regional Committees for Medical and Health Research Ethics, REC South-East C, 2019/ 28469, REK-13/05 and 2010/744). All data processing complies with the instructions of the Data Protection Authority in Iceland (PV_2017060950ÞS) and the Norwegian Data Inspectorate. Patients were involved in the design and conduct of several of the studies that are included in this report. Participants gave informed consent to participate in the study before taking part wherever applicable.
Acknowledgments
We would like to thank the individuals who participated in this study and the staff at the Icelandic Patient Recruitment Center, the deCODE genetics core facilities, the Swedish EIRA and EIMS study groups, the Swedish Rheumatology Quality Register Biobank study group (https://srq.nu/biobank-vardgivare/), KI Biobank at Karolinska Institutet, the Biobank Research Unit, Umeå University (https://www.umu.se/en/biobank-research-unit/), Västerbotten Intervention Programme, the Northern Sweden MONICA study and the County Council of Västerbotten for providing data and samples in Sweden; the Danish DANBIO registry and the Danish Rheumatologic Biobank for supplying data from Danish RA patients, including Niels Steen Krogh, Zitelab Aps, Denmark for database management. Further thanks to all our colleagues who contributed to the data collection and phenotypic characterisation of clinical samples, including Arni J Geirsson, Gudrun B Reynisdottir, Thorunn Jonsdottir and Gunnar Tomasson from Iceland, as well as Britt Corfixen and Tina M Kringelbach from Denmark. We also acknowledge colleagues working with the genotyping and analysis of the whole-genome association data. We would also like to thank Vibeke Østergaard Thomsen, International Reference Laboratory of Mycobacteriology, Statens Serum Institut and Marianne Kragh Thomsen Department of Clinical Microbiology, Aarhus University Hospital, Aarhus, Denmark, for collecting blood samples, as well as Elvira Chapka, Ewa Kogutowska and Mette Errebo Rønne, Statens Serum Institut, for laboratory support. We would like to thank the Norwegian Institute of Public Health for access to genomic data and the families in Norway who take part in the ongoing Norwegian Mother, Father and Child Cohort Study. Last but not least, we want to acknowledge the participants and investigators of the FinnGen study and the UK Biobank.
References
Footnotes
Handling editor Josef S Smolen
Collaborators Collaborators from the DBDS Genomic Consortium, the Danish RA Genetics Working Group and the Swedish Rheumatology Quality Register Biobank Study Group are listed in online supplemental information 1. Members of the DBDS Genomic Consortium: Steffen Andersen (Department of Finance Copenhagen Business School Copenhagen Denmark); Karina Banasik (Novo Nordisk Foundation Center for Protein Research Faculty of Health and Medical Sciences University of Copenhagen Copenhagen Denmark); Søren Brunak (Novo Nordisk Foundation Center for Protein Research Faculty of Health and Medical Sciences University of Copenhagen Copenhagen Denmark); Kristoffer Burgdorf (Department of Clinical Immunology Copenhagen University Hospital Copenhagen Denmark); Christian Erikstrup (Department of Clinical Immunology Aarhus University Hospital Aarhus Denmark); Thomas Folkmann Hansen (Danish Headache Center Department of Neurology Rigshospitalet Glostrup Denmark); Henrik Hjalgrim (Department of Epidemiology Research Statens Serum Institut Copenhagen Denmark); Gregor Jemec(Department of Clinical Medicine Zealand University Hospital Roskilde Denmark); Poul Jennum (Department of Clinical Neurophysiology at University of Copenhagen Copenhagen Denmark); Pär Ingemar Johansson (Department of Clinical Immunology Copenhagen University Hospital Copenhagen Denmark); Kasper Rene Nielsen (Department of Clinical Immunology Aalborg University Hospital Aalborg Denmark); Mette Nyegaard (Department of Biomedicine Aarhus University Denmark); Mie Topholm Brun (Department of Clinical Immunology Odense University Hospital Odense Denmark); Ole Birger Pedersen (Department of Clinical Immunology Zealand University Hospital, Køge Denmark); Susan Mikkelsen (Department of Clinical Immunology Aarhus University Hospital Aarhus Denmark); Khoa Manh Dinh (Department of Clinical Immunology Aarhus University Hospital Aarhus Denmark); Erik Sørensen (Department of Clinical Immunology Copenhagen University Hospital Copenhagen Denmark); Henrik Ullum (Department of Clinical Immunology Copenhagen University Hospital Copenhagen Denmark); Sisse Rye Ostrowski (Department of Clinical Immunology Copenhagen University Hospital Copenhagen Denmark); Thomas Werge (Institute of Biological Psychiatry Mental Health Centre Sct. Hans Copenhagen University Hospital Roskilde Denmark); Daniel Gudbjartsson (deCODE genetics Reykjavik Iceland); Kari Stefansson (deCODE genetics Reykjavik Iceland); Hreinn Stefánsson (deCODE genetics Reykjavik Iceland); Unnur Þorsteinsdóttir (deCODE genetics Reykjavik Iceland); Margit Anita Hørup Larsen(Department of Clinical Immunology Copenhagen University Hospital Copenhagen Denmark); Maria Didriksen (Department of Clinical Immunology Copenhagen University Hospital Copenhagen Denmark); Susanne Sækmose (Department of Clinical Immunology, Zealand University Hospital Køge Denmark). The Danish RA Genetics Working Group: Paal Skytt Andersen (Microbiology and Infection Control, Statens Serum Institut, Copenhagen, Denmark; Veterinary Disease Biology, University of Copenhagen, Copenhagen Denmark); Ram Benny Dessau (Department of Clinical Microbiology, Slagelse Hospital, Denmark); Malene Rohr Andersen (Department of Clinical Biochemistry, Herlev and Gentofte Hospital, University of Copenhagen, Hellerup, Denmark); Hans Jürgen Hoffmann (Department of Respiratory Diseases B, Institute for Clinical Medicine, Aarhus University Hospital, Aarhus, Denmark); Claus Lohman Brasen (Department of Biochemistry, Hospital of Lillebaelt, Vejle, Denmark). The Swedish Rheumatology Quality Register Biobank Study Group (SRQb): Johan Askling (Department of Medicine, Solna, Karolinska Institutet, Stockholm, Sweden); Eva Baecklund (Department of Medical Sciences, Section of Rheumatology, Uppsala University, Uppsala, Sweden); Lena Bjorkman (Department of Rheumatology and Inflammation research, Gothenburg University, Gothenburg, Sweden); Alf Kastbom (Department of Biomedical and Clinical Sciences, Linköping University, Linköping, Sweden); Solbritt Rantapaa-Dahlqvist (Reumatology, Section of Medicine, Department of Public Health and Clinical Medicine, Umea University, Umea, Sweden); Carl Turesson (Rheumatology, Department of Clinical Sciences, Malmö, Lund University, Malmö, Sweden).
Contributors SS, LS, PS, GT, UT, IJ and KS designed the study and interpreted the results. SS, BG, HW, GG, ICL, SBS, BAL, LA, EB, KB, SB, LB, TE, CE, OF, IG, OH, JH, EH, E-MH, SJ, DVJ, HJ, AK, IK, SK, HK, MHL, AL, AGL, TM, HM, TO, KH-P, HS, ES, IJS, CT, LAl, TKK, SB, KrS, VA, OAA, SR-D, MLH, LK, JA, OBP and IJ carried out the subject ascertainment and recruitment. SS, BG, HW, GG, ICL, SBS, BAL, MB, LA, KA, SB, CE, OF, IK, HK, BRL, TO, SRO, GNS, HS, ES, LA, TKK, SB, KrS, VA, OAA, SR-D, MLH, LK, JA, LP and OBP managed the data processing of participating study populations/biobanks. SS, LS, PS, EF, GR, AOA, DFG, SAG, GHH, SHL, GM, KHSM, PM, GLN, TAO, PIO, SR, UT and IJ performed the sequencing, genotyping, imputation, expression and proteomics analyses. SS, LS, PS, GT, EF, GR, SHL, TAO, DFG, PM, UT and IJ performed the statistical and bioinformatics analyses. SS, PS, GT, UT, IJ and KS drafted the manuscript. SS and KS accept full responsibility for the work, had access to the data and controlled the decision to publish. All authors contributed to the final version of the paper.
Funding The study was funded by NORDFORSK (grant agreement no. 90825, project NORA), the Swedish Research Council (2018-02803), the Swedish innovation Agency (Vinnova), Innovationsfonden and The Research Council of Norway, Region Stockholm-Karolinska Institutet and Region Västerbotten (ALF), the Danish Rheumatism Association (R194-A6956), the Swedish Brain Foundation, Nils and Bibbi Jensens Foundation, the Knut and Alice Wallenberg Foundation, Margaretha af Ugglas Foundation, the South-Eastern Heath Region of Norway, the Health Research Fund of Central Denmark Region, Region of Southern Denmark, the A.P. Moller Foundation for the Advancement of Medical Science, the Colitis-Crohn Foreningen, the Novo Nordisk Foundation (NNF15OC0016932), Aase og Ejnar Danielsens Fond, Beckett-Fonden, Augustinus Fonden, Knud and Edith Eriksens Mindefond, Laege Sofus Carl Emil Friis and Hustru Olga Doris Friis' Legat, the Psoriasis Forskningsfonden, the University of Aarhus, the Danish Rheumatism Association (R194-A6956, A1923, A3037 and A3570 – www. gigtforeningen.dk), Region of Southern Denmark’s PhD Fund, 12/7725 (www.regionsyddanmark.dk) and the Department of Rheumatology, Frederiksberg Hospital (www.frederiksberghospital.dk). MoBa Genetics has been funded by the Research Council of Norway (#229624, #223273), South East and Western Norway Health Authorities, ERC AdG project SELECTionPREDISPOSED, Stiftelsen Kristian Gerhard Jebsen, Trond Mohn Foundation, the Novo Nordisk Foundation and the University of Bergen. KB and SB acknowledge the Novo Nordisk Foundation (grant NNF14CC0001).
Competing interests Authors affiliated with deCODE Genetics/Amgen declare competing financial interests as employees. OAA is a consultant to HealthLytix. The following coauthors report the following but unrelated to the current report: Karolinska Institutet, with JA as principal investigator, has entered into agreements with the following entities, mainly but not exclusively for safety monitoring of rheumatology immunomodulators: Abbvie, BMS, Eli Lilly, Janssen, MSD, Pfizer, Roche, Samsung Bioepis and Sanofi, unrelated to the present study. SB has ownerships in Intomics A/S, Hoba Therapeutics Aps, Novo Nordisk A/S, Lundbeck A/S and managing board memberships in Proscion A/S and Intomics A/S. BG has received research grants from AbbVie, Bristol Myers-Squibb and Pfizer; OH has received research grants from AbbVie, Novartis and Pfizer, DVJ has received speaker and consultation fees from AbbVie, Janssen, Lilly, MSD, Novartis, Pfizer, Roche and UCB, AGL has received speaking and/or consulting fees from AbbVie, Janssen, Lilly, MSD, Novartis, Pfizer, Roche and UCB; and CT has received consulting fees from Roche, speaker fees from Abbvie, Bristol Myers-Squibb, Nordic Drugs, Pfizer and Roche, and an unrestricted grant from Bristol Myers-Squibb.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.