Article Text

Abstract
Objective To evaluate efficacy and safety of guselkumab, an anti-interleukin-23p19-subunit antibody, in patients with psoriatic arthritis (PsA) with prior inadequate response (IR) to tumour necrosis factor inhibitors (TNFi).
Methods Adults with active PsA (≥3 swollen and ≥3 tender joints) who discontinued ≤2 TNFi due to IR (lack of efficacy or intolerance) were randomised (2:1) to subcutaneous guselkumab 100 mg or placebo at week 0, week 4, then every 8 weeks (Q8W) through week 44. Patients receiving placebo crossed over to guselkumab at week 24. The primary (ACR20) and key secondary (change in HAQ-DI, ACR50, change in SF-36 PCS and PASI100) endpoints, at week 24, underwent fixed-sequence testing (two-sided α=0.05). Adverse events (AEs) were assessed through week 56.
Results Among 285 participants (female (52%), one (88%) or two (12%) prior TNFi), 88% of 189 guselkumab and 86% of 96 placebo→guselkumab patients completed study agent through week 44. A statistically significantly higher proportion of patients receiving guselkumab (44.4%) than placebo (19.8%) achieved ACR20 (%difference (95% CI): 24.6 (14.1 to 35.2); multiplicity-adjusted p<0.001) at week 24. Guselkumab was superior to placebo for each key secondary endpoint (multiplicity-adjusted p<0.01). ACR20 response (non-responder imputation) in the guselkumab group was 58% at week 48; >80% of week 24 responders maintained response at week 48. Through week 24, serious AEs/serious infections occurred in 3.7%/0.5% of 189 guselkumab-randomised and 3.1%/0% of 96 placebo-randomised patients; the guselkumab safety profile was similar through week 56, with no deaths or opportunistic infections.
Conclusion Guselkumab significantly improved joint and skin manifestations and physical function in patients with TNFi-IR PsA. A favourable benefit–risk profile was demonstrated through 1 year.
Trial registration number NCT03796858.
- arthritis
- psoriatic
- tumour necrosis factor inhibitors
- biological therapy
Data availability statement
The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Patients with psoriatic arthritis (PsA) with an inadequate response or intolerance to tumour necrosis factor inhibitors (TNFi) often have lower response rates to additional TNFi, and current treatment guidelines generally support only one switch within the TNFi class before selecting an alternate mechanism of action.
Guselkumab, a fully human interleukin (IL)−23 p19-subunit inhibitor, is efficacious in improving the signs and symptoms of active PsA both in TNFi-naïve and TNFi-experienced patients.
What does this study add?
In the phase III, randomised, placebo-controlled COSMOS study in adults with active PsA, guselkumab-treated patients had significantly higher response rates and greater mean improvements in assessments of the signs and symptoms of PsA at week 24 when compared with placebo; response rates and mean improvements were maintained or improved through 1 year in the guselkumab group.
The COSMOS safety results were consistent with the known safety profile of guselkumab in biologic-naïve patients with PsA.
How might this impact on clinical practice or future developments?
The efficacy and safety results of COSMOS suggest that guselkumab may be an appropriate therapy for patients with PsA with lack or efficacy from or intolerance to TNFi.
Introduction
Psoriatic arthritis (PsA) is a heterogeneous, chronic, inflammatory disease, with distinct classes of therapy now increasingly recommended based on the disease domains predominantly involved, such as enthesitis and dactylitis, in the individual patient.1 2 Current treatment guidelines3 recommend the use of a biologic disease-modifying antirheumatic drug (bDMARD) when conventional synthetic DMARDs (csDMARDs) have proven ineffective. The introduction of tumour necrosis factor inhibitors (TNFi) into the rheumatologist’s armamentarium has substantially improved the ability to achieve lower states of PsA activity3; however, up to 40% of patients receiving their first TNFi do not achieve response (assessed by ≥20% improvement in American College of Rheumatology criteria (ACR20)) with 6 months of treatment.3 An analysis of patients with PsA in the DANBIO registry who switched biologics after initiating TNFi therapy found decreased ACR20 response rates with the second and third TNFi (47%, 22% and 18%, respectively).4 In addition, real-world registry data have demonstrated diminished drug persistence with each successive TNFi.4–6
Alternate mechanisms of action may prove more beneficial in patients who experience a lack of response to TNFi,7 highlighting the need for treatments targeting alternate disease pathways. Accordingly, several bDMARDs with alternative mechanisms of action are now approved for PsA,8–10 including those targeting interleukin (IL)−17A, p40 (IL-12/23), and p19 (IL-23).
Guselkumab, a high-affinity, human monoclonal antibody targeting the IL-23p19-subunit, demonstrated efficacy and safety across two phase III PsA studies (DISCOVER-1 (TNFi-experienced and biologic-naïve), DISCOVER-2 (biologic-naïve only)).8 9 Approximately 31% of the 381 patients in DISCOVER-1 were previously exposed to 1–2 TNFi, and of those, 37% had discontinued TNFi therapy due to inadequate efficacy. The objective of the phase IIIb COSMOS study was to further assess the efficacy and safety of guselkumab through 1 year in patients with PsA with an inadequate response (IR; inadequate efficacy or intolerance) to TNFi.
Patients and methods
Patients
Eligible adults had a diagnosis of PsA according to the ClASsification criteria for Psoriatic ARthritis (CASPAR) at screening and had active disease (≥3 swollen; ≥3 tender joints) and active (≥1 psoriatic plaque of ≥2 cm) or documented history of plaque psoriasis or current nail psoriasis, and who had also demonstrated lack of benefit or intolerance to 1–2 TNFi. Patients could continue stable baseline use of methotrexate (MTX), sulfasalazine, hydroxychloroquine or leflunomide; oral corticosteroids; and non-steroidal anti-inflammatory drugs (NSAIDs)/other analgesics. Targeted synthetic DMARDs were prohibited before and during study participation. Patients with active tuberculosis (TB) were excluded; those with latent TB received appropriate prophylaxis.
Study design
This phase IIIb, randomised, double-blind study (COSMOS) was conducted at 84 European sites from March 2019 to November 2020 (see online supplemental methods). The study comprised a 6-week screening period and placebo-controlled (weeks 0–24) and active-treatment (weeks 24–48; final study intervention at week 44) periods. The primary endpoint assessment was at week 24, with final efficacy and safety assessments at week 48 and week 56, respectively.
Supplemental material
At week 0, participants were randomised (2:1) to receive subcutaneous injections of either guselkumab 100 mg (week 0, week 4, then every 8 weeks (Q8W) through week 44) or placebo (weeks 0, 4, 12, 20, followed by guselkumab 100 mg at weeks 24, 28, 36, 44). Randomisation was stratified by baseline csDMARD use (yes/no) and number of prior TNFi (1 or 2). Study personnel, including independent joint assessors and the study team, were blinded throughout the study. Participants with <5% improvement from baseline in both tender and swollen joint counts at week 16 qualified for early escape (EE); patients receiving guselkumab continued randomised treatment (receiving placebo at week 16 to maintain blinding), while those in the placebo group received guselkumab at week 16, week 20 and Q8W thereafter (figure 1). After EE, participants could initiate or increase the dose of one permitted concomitant medication up to the maximum allowed dose at the physician’s discretion. Sample size estimation is detailed in online supplemental methods.
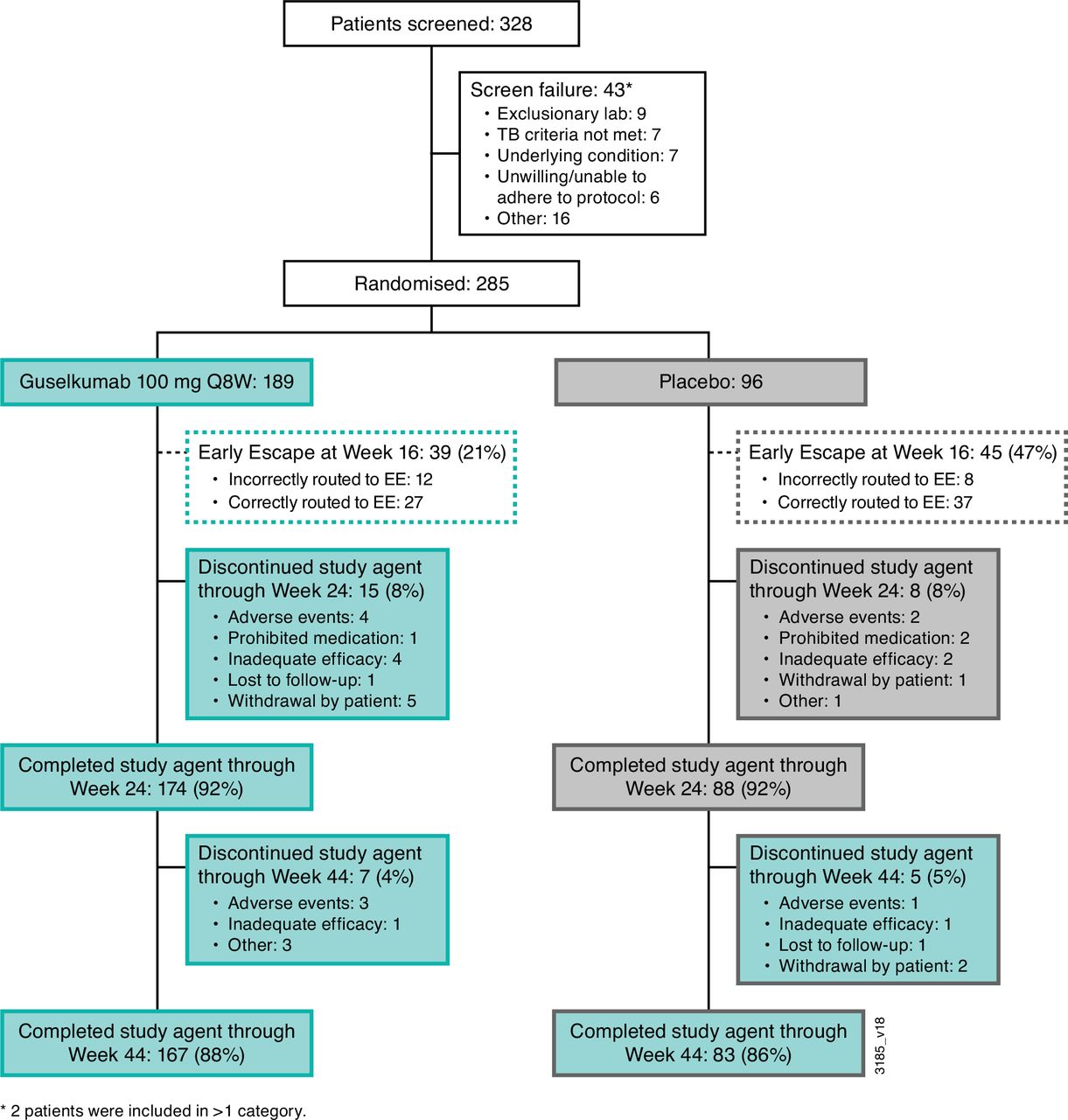
Disposition of patients through 1 year of COSMOS. EE, early escape; Q8W, every 8 weeks; TB, tuberculosis.
This study was conducted per the Declaration of Helsinki and Good Clinical Practice guidelines. Each site’s ethical body approved the protocol. Patients provided written informed consent.
Patient and public involvement statement
Patients and the public were not involved in the design or analysis of this study.
Procedures
Independent assessors evaluated joints for tenderness, swelling and presence/severity of enthesitis (Leeds Enthesitis Index (LEI))11 and dactylitis (Dactylitis Severity Score (DSS)).12 13 Patients reported pain and global arthritis activity (0–10 cm visual analogue scale (VAS)), and physical function (Health Assessment Questionnaire-Disability Index (HAQ-DI)).14 Investigators determined global disease activity (0–10 cm VAS), serum C reactive protein (CRP) and extent (% body surface area (BSA) with psoriasis) and severity of skin symptoms using the Investigator’s Global Assessment of psoriasis (IGA)15 and the Psoriasis Area and Severity Index (PASI).16 During the study, the protocol was amended to allow self-administration of study agent injections post-week 24 when site visits were not possible due to local COVID-19 restrictions.
The 36-item Short-Form Health Survey (SF-36) physical and mental component summary (PCS and MCS) scores assessed health-related quality of life (HRQoL).17 The Functional Assessment of Chronic Illness Therapy (FACIT)-Fatigue measured fatigue.18 Adverse events (AEs) and routine clinical laboratory parameters were monitored.
Outcomes
The primary endpoint was the proportion of patients with an ACR20 response at week 24. Major secondary endpoints, also at week 24, were (1) mean changes in HAQ-DI, (2) ACR50 response, (3) mean changes in SF-36 PCS and (4) PASI100 response (in patients with ≥3% BSA with psoriasis involvement and IGA ≥2 at baseline). Maintenance of ACR20/50/70 response at week 48 was also assessed in patients who achieved response at week 24. Additional secondary and safety outcomes assessed are shown in online supplemental methods.
Data analyses
Efficacy results were analysed by randomised treatment group, regardless of actual treatment received. The ‘Primary’ efficacy analysis included randomised participants who received ≥1 dose of study agent. Patients with missing data and those who met treatment failure (TF) criteria through week 24 (discontinued study agent and/or study participation for any reason, initiated or increased the dose of allowed csDMARDs or oral corticosteroids for PsA, initiated protocol prohibited medications/therapies for PsA or met EE criteria; online supplemental figure 1) were considered non-responders for binary endpoints or having no change for continuous endpoints (non-responder imputation (NRI)). Through week 24, least squares (LS) mean changes from baseline were determined for continuous endpoints using a Mixed-Effect Model Repeated Measures (MMRM) model including all available data through week 24 (additional details in online supplemental methods). Subgroup analyses evaluated consistency of the primary endpoint based on demographics, baseline disease characteristics and prior medications.
The overall type I error was controlled across the primary and major secondary endpoints at 5% by testing treatment differences (two-sided α=0.05) in a fixed sequence (ie, ACR20 response, change from baseline in HAQ-DI, ACR50 response, change from baseline in SF-36 PCS, PASI100 response; online supplemental figure 2), whereby subsequent endpoints were only tested if the previous endpoint achieved statistical significance (p<0.05). For endpoints not included in the multiplicity control procedure, the unadjusted (nominal) p values are descriptive in nature.
Supplemental sensitivity analyses, prespecified prior to the week 24 database lock, included a ‘Per-Protocol’ (PP) analysis (excluded patients with major protocol deviations (MPDs) with potential to impact efficacy assessments; online supplemental figure 3), and an ‘EE-Correction’ analysis (online supplemental figure 4). The latter analysis was conducted to address 20 patients (12 guselkumab, 8 placebo) incorrectly routed to EE and considered non-responders in the Primary analysis. In the EE-correction analysis, 12 affected patients in the guselkumab group did not meet any other TF criteria (eg, the introduction/change in dose of concomitant therapy) through week 24 and their response was included with those of other guselkumab-treated patients. The eight placebo patients received guselkumab as EE therapy at week 16 and week 20, thus met TF criteria, and were considered non-responders in the EE-correction analysis.
Through week 24, treatment group comparisons for binary endpoints used a Cochran–Mantel–Haenszel test stratified at the study level by baseline csDMARD use (yes/no) and number of prior TNFi (1/2) for binary endpoints or an MMRM model for continuous data (see online supplemental methods). Statistical analyses used SAS (V.9.4), with SAS/STAT (V.14.2; SAS Institute, Inc, Cary, NC, USA).
In post hoc analyses after week 24, results for the placebo→guselkumab group are reported for patients who crossed over to receive guselkumab at week 24. In addition, NRI was applied: patients who discontinued treatment and/or met EE criteria before week 24 (guselkumab group; excluding those who were incorrectly assigned to EE) were imputed as no response for binary endpoints or no change for continuous endpoints; missing data were imputed as no response or using multiple imputation (MI; assumed to be missing-at-random), respectively. After week 24, changes from baseline are reported as mean (SD). No treatment group comparisons were performed post-week 24.
Safety summaries included participants receiving ≥1 partial or complete administration of study agent, according to actual treatment received; numbers of events/100 patient-years (PY) of follow-up were determined for select AEs of interest.
Results
Patient disposition and characteristics
At week 0, 285 patients were randomised to guselkumab (n=189) or placebo (n=96); at week 16, 39 (21%) participants in the guselkumab group and 45 (47%) in the placebo group were assigned to EE. Through week 24, 15 (8%) and 8 (8%) participants, respectively, in the guselkumab and placebo groups discontinued study agent (figure 1). In total, 167 (88%) patients in the guselkumab group and 83 (86%) in the placebo-crossover group completed study treatment.
Although baseline characteristics were generally similar across treatment groups, several numerical imbalances existed, for example, a higher proportion of females and a lower mean body weight in the guselkumab (54%, 84 kg) than placebo (46%, 92 kg) group. The guselkumab group was characterised by more prominent joint symptoms (tender joint count: 21 vs 18) and skin involvement (mean PASI: 11.7 vs 9.2). Prior and concomitant medications were similar across groups (table 1).
Baseline characteristics of COSMOS participants
Although self-administration was permitted during the COVID-19 pandemic, when site visits were restricted, MPDs related to COVID-19 did occur. These were classified mostly as drug administration or study visit missed or outside of the prespecified window, and most were considered to have no effect on efficacy assessments.
Efficacy
The primary endpoint was met. At week 24, based on the Primary analysis population (online supplemental figure 1), 44.4% (84/189) of guselkumab versus 19.8% (19/96) of placebo patients achieved ACR20 (%difference (95% CI): 24.6 (14.1 to 35.2); multiplicity-adjusted p<0.001), with treatment effect seen by week 4 (figure 2A). Results of the PP and EE-correction sensitivity analyses supported the Primary analysis. Specific to the EE-correction analysis, 48.1% (91/189) of guselkumab versus 19.8% (19/96) of placebo patients achieved ACR20 (%difference (95% CI): 28.2 (17.7 to 38.8)) (figure 2B). The benefit of guselkumab over placebo was consistent across subgroups defined by baseline patient, disease and prior/concomitant medication characteristics, including participants who discontinued prior TNFi use due to inadequate efficacy or intolerance (figure 3). Employing NRI, the proportion of guselkumab-randomised patients achieving ACR20 at week 48 was 57.7%. Among 51 placebo patients who crossed over to guselkumab at week 24, 54.9% (n=28) achieved ACR20 at week 48 (figure 2A).

ACR20 response through week 48 of COSMOS. Proportions of randomised and treated patients achieving ACR20 response through week 24 in the Primary analysis (treatment failure rules applied) (A) and ACR20 response at week 24 across the Primary, PP and EE-correction analyses (B). After week 24, analyses were performed using non-responder imputation methods, including imputation of EE patients as non-responders (see Patients and methods). Results for the placebo→guselkumab group at week 48 are reported for patients who did not enter EE and crossed over to guselkumab at week 24. ACR20, ≥20% improvement in American College of Rheumatology response criteria; EE, early escape; GUS, guselkumab; PBO, placebo; PP, per protocol; Q8W, every 8 weeks.
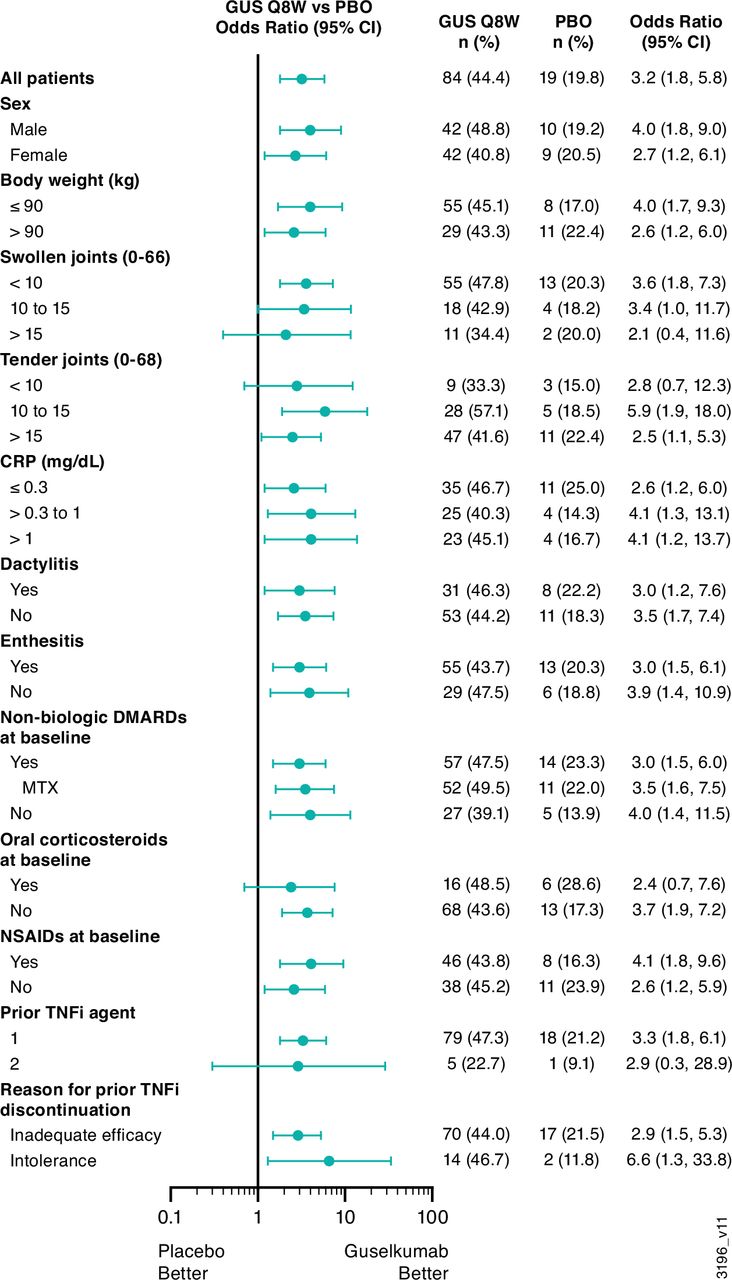
ACR20 response at week 24 by baseline characteristics of COSMOS participants. ACR20, ≥20% improvement in American College of Rheumatology response criteria; CRP, C reactive protein; DMARD, disease-modifying antirheumatic drug; GUS, guselkumab; MTX, methotrexate; NSAID, non-steroidal anti-inflammatory drug; PBO, placebo; Q8W, every 8 weeks; TNFi, tumour necrosis factor inhibitor.
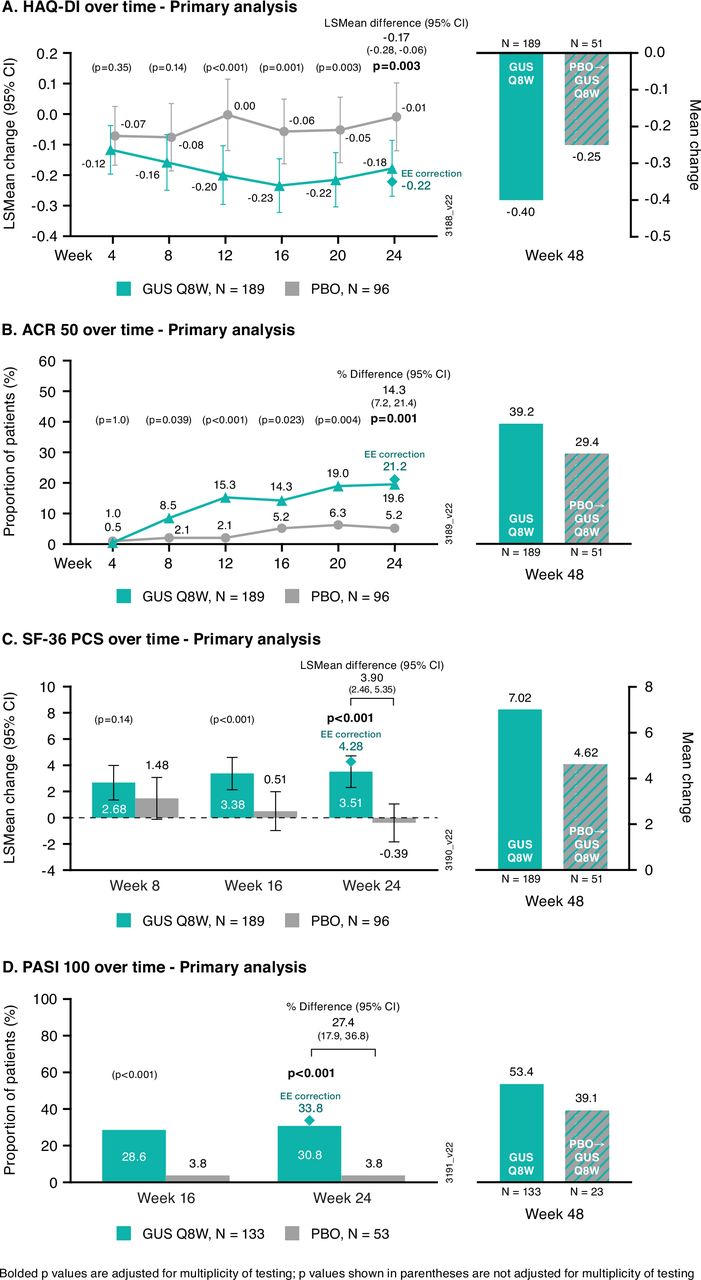
The testing hierarchy did not fail in analyses of the major secondary endpoints; guselkumab was superior to placebo in all four endpoints. At week 24, guselkumab patients demonstrated statistically significantly greater improvements or response rates versus placebo in HAQ-DI score (LSmean (95% CI) change: −0.18 (−0.27 to –0.09) vs −0.01 (−0.12 to 0.10); multiplicity-adjusted p=0.003; figure 4A), ACR50 (19.6% (37/189) vs 5.2% (5/96); multiplicity-adjusted p=0.001; figure 4B), SF-36 PCS score (LSmean (95% CI) change: 3.51 (2.31 to 4.72) vs −0.39 (−1.84 to 1.07); multiplicity-adjusted p<0.001; figure 4C) and PASI100 (in patients with ≥3% BSA with psoriasis and IGA ≥2 at baseline; 30.8% vs 3.8%; multiplicity-adjusted p<0.001; figure 4D). Results of PP and EE-correction sensitivity analyses were consistent with the Primary analysis (Supplemental Figure 5A−D).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Key secondary outcomes through week 48 of COSMOS. Primary analysis through week 24 and post hoc NRI analysis at week 48 of LSmean change and mean change in HAQ-DI score (A), ACR50 response (B), LSmean change and mean change in SF-36 PCS score (C), and PASI100 response (D). After week 24, analyses were performed using NRI (including imputation of EE patients as non-responders in the guselkumab group; see Patients and methods). Results for the placebo→guselkumab group at week 48 are reported for patients who did not enter EE and crossed over to guselkumab at week 24. ACR50, ≥50% improvement in American College of Rheumatology response criteria; GUS, guselkumab; HAQ-DI, Health Assessment Questionnaire-Disability Index; LS, least squares; NRI, non-responder imputation; PASI100, 100% improvement in Psoriasis Area and Severity Index; PBO, placebo; Q8W, every 8 weeks; SF-36 PCS, 36-item Short-Form Health Survey Physical Component Summary.
Additional secondary endpoints at week 24 also showed benefit of guselkumab over placebo for achieving ACR70 (7.9% vs 1.0%; nominal p=0.018), minimal disease activity (MDA; 14.8% vs 3.1%; nominal p=0.003), and PASI75 (59.4% vs 9.4%; nominal p<0.001) and PASI90 (51.1% vs 7.5%; nominal p<0.001) in patients with ≥3% BSA with psoriasis and IGA ≥2 at baseline; 3.7% guselkumab-treated and no placebo-treated patients achieved very low disease activity. At week 24, guselkumab-treated patients also had a greater LSmean change in Disease Activity in Psoriatic Arthritis (DAPSA) score (−14.5 vs −5.7; nominal p<0.001) and a higher DAPSA low disease activity (LDA) response rate (29.6% vs 13.5%, nominal p=0.003) versus placebo; the proportion of patients achieving DAPSA remission was numerically higher in the guselkumab group versus placebo (5.3% vs 2.1%). Among participants affected at baseline, numerically higher proportions of guselkumab than placebo patients had resolved enthesitis (39.7% vs 18.8%; nominal p=0.003) or dactylitis (44.8% vs 25.0%; nominal p=0.040) at week 24. Guselkumab-treated patients also had greater LSmean improvements across all ACR components compared with placebo (Supplemental Figure 6A−G). The LSmean changes in SF-36 MCS were 2.10 and 0.36, respectively, in the guselkumab and placebo groups (table 2). In addition, higher proportions of guselkumab than placebo patients achieved clinically meaningful improvements in HAQ-DI (37.5% vs 16.1%; nominal p<0.001; table 2), FACIT-F (42.9% vs 20.8%; nominal p<0.001; table 2), and SF-36 PCS (42.3% vs 15.6%; nominal p<0.001) and MCS (28.6% vs 15.6%; nominal p=0.016) scores.
Additional secondary efficacy assessments at week 24 and week 48 analysed using non-responder imputation*
After week 24, response rates and mean improvements for secondary endpoints were sustained or numerically improved through week 48 in guselkumab-randomised patients (figure 4A–D and table 2). Among placebo→guselkumab patients, response rates and mean changes in the secondary endpoints increased at week 48 (figure 4A–D and table 2).
Maintenance of response was evaluated for guselkumab-randomised patients achieving an ACR20, ACR50 or ACR70 response at week 24; of these patients, 83.3% (70/84), 81.1% (30/37) and 86.7% (13/15), respectively, maintained response at week 48.
Safety
Through week 24, similar proportions of patients in the guselkumab (42% (80/189)) and placebo (48% (46/96)) groups reported ≥1 AE. Through week 56, 144.9 AEs/100PY were reported among the 279 guselkumab-treated patients (vs 369.8/100PY for placebo). The most common AEs in guselkumab-randomised patients through week 24, ie, nasopharyngitis (5%) and upper respiratory tract infection (4%), occurred with similar incidence in the placebo group (5% and 3%, respectively) (table 3). Infections remained the most common AEs in guselkumab-treated patients through week 56 (37.2/100PY vs 99.6/100PY for placebo).
Summary of adverse events through week 56 of COSMOS
The incidences of serious AEs (SAEs) and AEs leading to treatment discontinuation were 6.3/100PY and 2.7/100PY, respectively, among guselkumab-treated patients through week 56. One patient experienced a major adverse cardiovascular event at week 44 (non-fatal myocardial infarction (preferred term: acute coronary syndrome)); risk factors included concomitant cyclooxygenase-2-inhibitor therapy and a body mass index of 31. One malignancy occurred: prostatic adenocarcinoma in a guselkumab-randomised patient (4-year history of prostatitis). One patient discontinued study agent (influenza-like illness) after the third guselkumab administration and was diagnosed with suspected inflammatory bowel disease and coeliac disease ~3 weeks and 2 months, respectively, later. Neither diagnosis was confirmed; the patient was lost to follow-up.
One patient (guselkumab group) experienced two events of conversion disorder, requiring hospitalisation; study drug was discontinued after the second instance, which was reported as resolved. Another patient in the guselkumab group (history of previous suicide attempt) reported depression (SAE) 1 week after receiving the second guselkumab administration; study agent was discontinued, with no further follow-up. Other non-serious psychiatric-related AEs were singular events of anxiety and depressed mood in the placebo group (through week 24) and insomnia in the guselkumab group.
Two serious infections occurred. One guselkumab-randomised patient was hospitalised with community-acquired pneumonia diagnosed at week 12 (history of chronic obstructive pulmonary disease and heart disease); the patient recovered with antibiotic treatment and resumed study agent. A placebo→guselkumab patient was hospitalised with acute pneumonia (week 48); the patient recovered following antibiotic therapy and continued study participation. No opportunistic infections, cases of active TB, or deaths occurred (table 3).
Injection-site reactions, all considered of mild intensity, occurred in 1.8% of guselkumab-treated patients (table 3). No anaphylactic or serum sickness-like reactions occurred through week 56.
Through week 56, AEs of decreased neutrophil and white blood cell counts were uncommon. Neither type of haematological abnormality was reported as an SAE or led to study agent discontinuation, and all were National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) Grade ≤2 (online supplemental table 1). The majority of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) elevations were maximum NCI-CTCAE Grade 1 (online supplemental table 1). Two guselkumab-treated patients had elevated ALT reported as an SAE. The first patient, whose liver enzymes were elevated at baseline, was confirmed to have autoimmune hepatitis via biopsy and imaging studies and discontinued study agent. While ALT levels normalised by week 24, other symptoms (jaundice, nausea) persisted. A second patient had elevated AST and ALT at week 48 and was diagnosed with steatohepatitis; the patient was treated with ademetionine and recovered. ALT and AST elevations occurred in 37% and 28%, respectively, in patients receiving concomitant MTX and in 28% and 24% of those not receiving concomitant MTX.
Discussion
Guselkumab-treated patients had statistically significant improvements in the signs and symptoms of PsA in TNFi-IR patients compared with placebo. The primary endpoint was achieved (ACR20: guselkumab, 44% vs placebo, 20%). Guselkumab 100 mg Q8W afforded higher ACR20 and ACR50 response rates, as early as week 4 and week 8, respectively. Furthermore, >80% of patients who achieved ACR20/50/70 at week 24 maintained response at week 48. In addition, this study demonstrated the efficacy of guselkumab in resolving enthesitis and dactylitis, achieving clear skin and achieving MDA in patients with TNFi-IR PsA. The guselkumab group also had greater improvements in fatigue, physical function and HRQoL scores than placebo at week 24, with approximately 30%–40% of guselkumab-randomised patients achieving an improvement greater than the minimal clinically important differences at week 24.
Importantly in this TNF-IR population, improvements in signs and symptoms of PsA were maintained or numerically increased through week 48 among guselkumab-randomised patients. Among placebo→guselkumab patients, response rates and mean improvements increased through week 48. Thus, guselkumab 100 mg Q8W demonstrated efficacy through 1 year across the diverse symptoms in patients with TNFi-IR PsA.
Prespecified sensitivity analyses (eg, excluding patients with MPDs relevant to efficacy outcomes and correcting errors in EE patients thus providing a more accurate assessment of treatment effect) confirmed those of the primary endpoint (ACR20 at week 24). Although absolute response rates tended to be numerically lower in COSMOS patients relative to the primarily biologic-naïve populations in previous studies, the treatment effect of guselkumab as measured by the difference between the Q8W group and placebo at week 24 (ACR20 %differences: 25─28% across primary and sensitivity analyses) was generally consistent with that observed for guselkumab 100 mg Q8W in largely biologic-naïve patients with active PsA in the similarly designed pivotal DISCOVER-1 and DISCOVER-2 studies (ACR20 %differences: 30─31%).8 9
Guselkumab was well tolerated by participants, demonstrating a safety profile similar to placebo. Two guselkumab-treated patients had a serious infection. Two placebo-treated patients and three guselkumab-treated patients reported psychiatric disorders; two were SAEs, one occurring in a patient with a prior history of suicide attempt. One case of suspected, but unconfirmed, inflammatory bowel disease was reported ~1 month after the patient discontinued guselkumab due to an influenza-like illness. Abnormal clinical laboratory findings were uncommon; no participant died or developed an opportunistic infection or TB. Thus, these safety findings in patients with TNFi-IR PsA through week 56 of COSMOS expand on, and are consistent with, the accumulated guselkumab safety profile established in patients with psoriasis receiving guselkumab through 5 years19 and that seen in DISCOVER-1 (1 year)20 and DISCOVER-2 (2 years).21 22
Although head-to-head trials comparing guselkumab with other targeted or biologic therapies have not been conducted in patients with PsA, results from a recent network meta-analysis found that guselkumab had comparable efficacy with TNFi and IL-17A inhibitors in achieving ACR response in biologic-naïve patients.23 In addition, the rates of AEs and SAEs were generally similar across treatment modalities; however, comparisons were limited by the significant uncertainty in the comparisons.23 It is generally recommended to switch to an alternate mechanism of action following biologic treatment failure,1 2 with only one switch between TNFi now recommended by the European Alliance of Associations for Rheumatology.2 The demonstrated efficacy of therapies targeting the IL-12/23p40-subunit, IL-17A, and Janus kinases in TNFi-experienced patients with PsA24–27 further supports the use of novel therapies to target alternative disease pathways.
However, patients who have experienced IR with a biologic, such as those enrolled in COSMOS, are at continued risk of treatment failure with subsequent therapies, thus highlighting the recalcitrant nature of the disease course in some patients with PsA.4–6 Of note, 88% of guselkumab-randomised patients in COSMOS remained on treatment, and 94% of placebo patients who received guselkumab after week 24 completed study treatment through week 44. High study retention in COSMOS may thus reflect a positive benefit–risk profile for patients who had inadequate response to previous TNFi therapy. Patients who did not achieve an ACR20 response may have experienced substantial improvement in other symptoms (eg, skin disease). Other factors, such as comorbidities and limited availability or concerns about adverse effects of alternative treatment options in this refractory population, may have also contributed to patient retention.
Numerical imbalances in baseline characteristics (eg, gender, weight, joint counts and severity of skin disease) and errors in EE assignment may have influenced efficacy, although predominately not in favour of guselkumab. The slight imbalance between the treatment groups in the proportion of women is noteworthy considering research demonstrating that among patients with PsA, women tend to report having a higher disease burden and lower levels of response to treatment compared with men.28 In addition, a separate analysis of 855 patients with PsA treated at a single rheumatology clinic found that being overweight was associated with not achieving treatment goals, specifically for women; however, no information was provided on the specific treatments these patients received.29
The COSMOS study was conducted across Europe, limiting ethnic diversity. COVID-related regulations during the latter half of study conduct may have increased MPDs; however, most were related to timing of study visits and did not impact efficacy. While the positive guselkumab benefit–risk profile observed through week 24 was maintained through 1 year, real-world evidence will further inform long-term guselkumab persistence in TNFi-IR patients.
In conclusion, guselkumab 100 mg Q8W was effective in patients with TNFi-IR PsA and demonstrated a favourable benefit–risk profile through 1 year. The statistically significant improvements observed with guselkumab across multiple clinical disease domains suggest a broad impact of targeting the p19 subunit of IL-23 in TNFi treatment-resistant PsA.
Data availability statement
The data sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
Ethics statements
Patient consent for publication
Ethics approval
The governing ethical bodies for each of the 84 participating sites approved the COSMOS study protocol.
Acknowledgments
The authors thank Cynthia Guzzo, MD (consultant funded by Janssen) for assistance with manuscript preparation; Soumya D Chakravarty, MD PhD, (Janssen Scientific Affairs, LLC employee) for substantive manuscript review; Michelle L Perate, MS, and Rebecca Clemente, PhD (Janssen Scientific Affairs, LLC employees) for assistance with manuscript preparation and submission; and Wenzhong Tang, MS (Janssen Research & Development, LLC employee) for statistical support. Some of these data were presented at EULAR 2021.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
Contributors Substantial intellectual contribution to conception and design, or acquisition of data, or analysis and interpretation of data: LCC, LG, ET, PB, MN, CSK, MS, WN, GS, IBM. Drafting the article or revising it critically for important intellectual content: LCC, LG, ET, PB, MN, CSK, MS, WN, GS, IBM. Final approval of the version to be published: LCC, LG, ET, PB, MN, CSK, MS, WN, GS, IBM. LCC is the guarantor.
Funding Janssen Research & Development, LLC funded this study.
Competing interests LCC has received consulting fees from AbbVie, Amgen, Biogen, BMS, Boehringer Ingelheim, Celgene, Domain, Eli Lilly, Gilead, Janssen, Medac, Novartis, Pfizer and UCB. LCC is funded by a National Institute for Health Research Clinician Scientist award. The work was supported by the National Institute for Health Research (NIHR) Oxford Biomedical Research Centre (BRC). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. LG has received research grants from Amgen, Eli Lilly, Galapagos, Janssen, Pfizer, Sandoz, and consulting fees from AbbVie, Amgen, Biogen, Bristol Myers Squibb, Celgene, Eli Lilly, Galapagos, Gilead, Janssen, Novartis, Pfizer, Samsung Bioepis, Sanofi-Aventis and UCB. ET is an employee of Janssen Scientific Affairs, LLC. PB, MN and WN are employed by Janssen Scientific Affairs, LLC (a subsidiary of Johnson & Johnson) and own Johnson & Johnson stock and/or stock options. CSK is employed by Janssen Research & Development, LLC (a subsidiary of Johnson & Johnson) and owns Johnson & Johnson stock and/or stock options. MS is employed by Immunology Global Medical Affairs, Janssen Pharmaceutical Companies of Johnson & Johnson and owns stock in Johnson & Johnson. GS has received speaker’s honoraria from Amgen, AbbVie, Bristol Myers Squibb, Eli Lilly, Gilead, Janssen, Novartis and UCB. IBMcI has received research grants and/or honoraria (all <$10 000 per annum) from AbbVie, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Eli Lilly, Janssen, Novartis, Pfizer and UCB.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.