Article Text

Abstract
Objectives To characterise splicing machinery (SM) alterations in leucocytes of patients with rheumatoid arthritis (RA), and to assess its influence on their clinical profile and therapeutic response.
Methods Leucocyte subtypes from 129 patients with RA and 29 healthy donors (HD) were purified, and 45 selected SM elements (SME) were evaluated by quantitative PCR-array based on microfluidic technology (Fluidigm). Modulation by anti-tumour necrosis factor (TNF) therapy and underlying regulatory mechanisms were assessed.
Results An altered expression of several SME was found in RA leucocytes. Eight elements (SNRNP70, SNRNP200, U2AF2, RNU4ATAC, RBM3, RBM17, KHDRBS1 and SRSF10) were equally altered in all leucocytes subtypes. Logistic regressions revealed that this signature might: discriminate RA and HD, and anti-citrullinated protein antibodies (ACPAs) positivity; classify high-disease activity (disease activity score-28 (DAS28) >5.1); recognise radiological involvement; and identify patients showing atheroma plaques. Furthermore, this signature was altered in RA synovial fluid and ankle joints of K/BxN-arthritic mice. An available RNA-seq data set enabled to validate data and identified distinctive splicing events and splicing variants among patients with RA expressing high and low SME levels. 3 and 6 months anti-TNF therapy reversed their expression in parallel to the reduction of the inflammatory profile. In vitro, ACPAs modulated SME, at least partially, by Fc Receptor (FcR)-dependent mechanisms. Key inflammatory cytokines further altered SME. Lastly, induced SNRNP70-overexpression and KHDRBS1-overexpression reversed inflammation in lymphocytes, NETosis in neutrophils and adhesion in RA monocytes and influenced activity of RA synovial fibroblasts.
Conclusions Overall, we have characterised for the first time a signature comprising eight dysregulated SME in RA leucocytes from both peripheral blood and synovial fluid, linked to disease pathophysiology, modulated by ACPAs and reversed by anti-TNF therapy.
- rheumatoid arthritis
- anti-citrullinated protein antibodies
- biological therapy
- tumor necrosis factor inhibitors
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. It would be also available from the corresponding author upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
- rheumatoid arthritis
- anti-citrullinated protein antibodies
- biological therapy
- tumor necrosis factor inhibitors
Key messages
What is already known about this subject?
Although there is recent evidence demonstrating the relevance of alternative splicing in tumorous and inflammatory pathologies, and some studies have shown association between the presence of splice variants and clinical profile of patients with rheumatoid arthritis (RA), alterations of the splicing machinery and their involvement in this disease have not been analysed so far.
What does this study add?
A signature comprising eight dysregulated splicing machinery elements (SME) has been identified in RA leucocytes subsets from peripheral blood, linked to key clinical features of this disease.
SME are further altered in mononuclear cells from RA synovial fluid, synovial tissue and ankle joints of K/BxN-arthritic mice and modulated in vivo by anti-tumour necrosis factor therapy.
Mechanistic studies have identified underlying mechanisms promoting SME alteration, involving both inflammatory mediators and autoantibodies (anti-citrullinated protein antibodies). Moreover, the reversion of their aberrant expression levels ameliorated the pathogenic RA phenotype of immune cells and synovial fibroblasts.
How might this impact on clinical practice or future developments?
The characterisation of new molecular mechanisms associated with the pathogenesis of RA, such as the presence of altered SME, might drive the development of potential biomarkers of disease and new therapeutic avenues for the management of this and other related immune-mediated disorders.
Introduction
Rheumatoid arthritis (RA) is a systemic autoimmune disease characterised by polyarthritis, joint damage and functional disability. Patients with RA exhibit increased frequency of cardiovascular disease, higher susceptibility to infections and increased risk for certain malignancies.1 Complex networks of pro-inflammatory cytokines, chemokines and growth factors play a fundamental role in its pathogenesis. Nevertheless, patients with RA display high heterogeneity in their clinical evolution and response to therapy, so that the precise mechanisms underlying the pathophysiology of the disease need further elucidation.
The onset of RA seems to be triggered by genetic environmental interactions that foster autoimmunity, based on genetic predisposition combined with repeated activation of the innate and adaptive immune systems.1 Genetic analyses have identified specific loci associated to RA onset and/or related comorbidities.2 Besides, a considerable number of genes differentially expressed on several cell populations have been characterised as predictors of clinical evolution and therapeutic response.3 4 Epigenetic studies based on DNA methylation5 and microRNAs6 have also provided novel mechanisms underlying the RA pathogenesis. Despite these findings, the information generated by genomic analyses is incomplete and shows certain limitations.
Gene transcription is tightly coupled to the subsequent splicing process, whereby introns are excised and exons are pasted together in mature RNAs by an intricate nuclear molecular machinery, the spliceosome, which consist on a discrete set of ribonucleoproteins and proteins, aided by more than 300 splicing factors.7 Alternative splicing may generate different mature RNA arising from the same gene, which precisely defines the final quantitative outcome of gene expression and impacts the functional diversification of proteins. Although the role of alternative splicing in RA has received limited attention to date, some studies have shown association between the presence of splice variants and clinical features of RA. Events related to alternative splicing previously reported in RA include the presence of splice variants of adhesion molecules such as fibronectin (FN1) in the microvasculature of the synovium,8 9 proangiogenic factors (VEGF and CXCL12), on the synovial tissue,10 regulators of cell transcription (FOXP3) in synovial lymphocytes11 and on synovium fibroblasts (TNFAIP3, BRAF and BIRC5),12 13 as well as genes involved in adhesion and cell metabolism in peripheral mononuclear cells (CD44 and MAP2K4),14 15 and monocytes (STEAP4).16 As well, increased circulating levels of protein isoforms generated by alternative splicing, such as TNFR2, PTPN22, SELE, ILR6 and ILR7 have been also demonstrated in RA.17–21 Most of these events have been associated with diverse RA features, thus supporting that the RNA splicing process might be severely altered in these autoimmune patients.
Nevertheless, alterations of the splicing machinery (SM) and their involvement in RA disease have not been analysed so far. Thus, the aim of this study was to explore and characterise the potential alterations of the SM components in peripheral blood leucocytes of patients with RA, and to define their influence on disease activity, its inflammatory and atherothrombotic profiles and the response to therapy.
Our results identified, for the first time, a signature comprising eight altered SM components in RA leucocytes, associated with key clinical features and therapy effectiveness.
Patients and methods
One hundred patients with RA and 29 healthy donors (HD) were included in the study (during a 24-month period), and involving two patients’ cohorts. The first cohort comprised 72 patients with RA and 29 HD, whose clinical and laboratory details are displayed in table 1. All patients with RA fulfilled the American College of Rheumatology criteria for the classification of RA.22 Patients and HD provided written informed consent. None of the HD had a history of other autoimmune diseases, atherosclerosis or thrombosis. The second cohort consisted of 38 patients with RA treated with anti-tumour necrosis factor (TNF)α drugs (TNF inhibitor (TNFi)) at standard dosage for 3 and 6 months. TNFi response was assessed by European League Against Rheumatism criteria.23 The study was conducted in accordance with the Declaration of Helsinki principles.
Clinical details of the patients with rheumatoid arthritis and healthy donors recruited to the study
Blood sample collection, assessment of clinical and biological parameters and B-mode ultrasound IMT measurements (see online supplemental materials).
Supplemental material
Analysis of SM components by qPCR microfluidic Dynamic Array
A 48.48 Dynamic Array (Fluidigm) was used to assess the expression of 45 selected transcripts of the major and minor spliceosome and associated splicing factors as previously reported.24–28 Briefly, an ‘integrated fluidic circuit’ (IFC) is connected to reagent input wells to perform quantitative PCR (qPCR, which is detected by fluorescence.
Separated tests were developed for each cohort of patients previously detailed, including, respectively, 72 and 38 patients with RA, and 29 HD (see online supplemental materials for further details).
RNAseq analysis of public data set to gain insight in the splicing alteration
RNA-seq data of an external cohort of 44 patients (E-MTAB-6141)29 was analysed as an independent cohort to explore enriched gene pathways, splicing variants and several splicing events associated to the dysregulation of the SM, as well as to validate the differential alteration of synovium and blood. The study of splicing variants and events was performed using Salmon, DESeq and SUPPA2 softwares30–32 which allowed to assess the relative abundances of the splicing events as Percent Spliced In Index (PSI or Ψ) and explore their association with gene expression levels (low/high expression) (see online supplemental materials).
Bio-Plex assay of the inflammatory profile
Secreted levels of cytokines/chemokines/adhesion molecules in plasma of the two cohorts of 72 and 38 patients with RA, respectively, were determined using a 27-plex panel in a multiplex bead-based assay system (Bio-Plex multiplex immunoassays, Bio-Rad; California, USA) (see online supplemental materials).
SM analysis in K/BxN mice
Arthritis was induced in 6–8 week-old mice by intraperitoneal injection of 100 µl of K/BxN (KRNxNOD) serum on days 0 and 2. Characteristic of mice, arthritis induction and microarray analysis have been previously described.33 Briefly, total RNA was obtained from ankle joints of three male arthritic mice and three control, non-arthritic mice. The joints were taken 7 days after serum transfer and immediately frozen in liquid nitrogen. To this end, hind limbs were prepared by dissecting the skin and muscle, and then sectioning ankle joints. Genome-wide microarray analysis was performed with the Mouse Gene 1.0 ST array (Affymetrix, Santa Clara, California, USA) at Progenika BioPharma SA (Bilbao, Spain) and SM components were identified.
In vitro studies
Four sets of in vitro experiments were developed to interrogate mechanistically the role of the altered SM in RA:
Treatment of HD leucocytes subsets with IgG-anti-citrullinated protein antibodies (ACPAs) or IgG-depleted ACPAs—purified from serum of active patients with RA—to evaluate their effects on the expression of both, the eight commonly altered SM elements (SME) and several inflammatory mediators. In addition, these treatments were also performed in the presence of FcR blocking.
Treatment of HD leucocytes subsets with key cytokines linked to the pathophysiology of RA (TNFα, interleukin (IL)-6 and CCL2), in order to assess their involvement in the aberrant expression of the eight commonly altered SME.
Transfection studies with KHDRBS1 and SNRNP70 in RA purified leucocytes subsets, to evaluate changes in cell activity promoted by overexpression of these SM components.
Treatment of RA-purified synovial fibroblasts (SF) with supernatants of the KHDRBS1 and SNRNP70-transfected lymphocytes, to analyse the functional consequences of SME modulation (see online supplemental materials for further details).
Cultured neutrophils were treated for 6 hours, and monocytes, lymphocytes and SF were treated for 24 hours before the respective analysis.
Identification of the citrullinome in PBMCs by LC-MS/MS
Citrullinome was evaluated by liquid chromatography-tandem mass spectrometry (LC-MS/MS) in pooled cell lysates from peripheral blood mononuclear cells (PBMCs) isolated from two sets of five patients with RA each, including those with severe SME alteration (low expression levels) and those with mild SME alteration (high expression levels). Mass spectrometry raw files were processed with PEAKS Studio 10.6 build 20201221 (Bioinformatics Solutions). For post-translational modification quantification, citrullinated peptides with AScore >20 (p value<0.01) were considered34 (see online supplemental materials for further details).
Statistical analyses
Data were expressed as mean±SEM or median±IQR according to data distribution, evaluated using Kolmogorov-Smirnov test. Student’s t-test or Mann-Whitney rank-sum test were used to assess statistical differences in unpaired data, and paired t-tests and Wilcoxon matched-pairs signed-rank tests for paired data. The χ2 test was used to associate qualitative variables. Correlations were evaluated by Spearman’s correlation test. Statistically significant differences were considered at p value<0.05 and false discovery rate (FDR)<0.15.
Logistic regression models and receiver operating characteristic curves were performed to evaluate the specificity and sensibility of the different discriminating models (see online supplemental materials for further details).
Results
The SM is profoundly altered in RA peripheral blood leucocytes
Twenty-one components of the SM out of the 45 total analysed were found differentially expressed in RA monocytes (figure 1A). All of them, including major and minor spliceosome components and splicing factors, were found reduced in monocytes from patients with RA. Remarkably, only RNU4ATAC, a key component of the minor spliceosome,35 was overexpressed. Similarly, 14 components of the SM were differentially expressed in lymphocytes from RA (figure 1B). All of them were found reduced and, in line with monocytes, RNU4ATAC and NOVA1 were found overexpressed. Twenty-three components of the SM were differentially expressed in neutrophils from patients with RA (figure 1C). Similarly, most of them were found reduced, being overexpressed RNU4ATAC, NOVA1 and CELF1.

Splicing machinery is highly altered in leucocytes from patients with RA. Expression levels of major and minor spliceosome and associated splicing factors were quantified through a 48.48 Dynamic Array (Fluidigm) in monocytes (A), lymphocytes (B) and neutrophils (C) from 29 peripheral blood of healthy donors (HD) and 72 patients with rheumatoid arthritis (RA). Heat map are displayed on top of each panel showing differential expression in the splicing machinery between RA and HD (log10 fold change). Blue and red colours represent downregulated and upregulated splicing machinery elements, respectively, while those showing significant differences (p<0.05) are highlighted in bold. Violin plots are also displayed at the bottom of each panel representing the expression levels of the differentially expressed spliceosome and splicing factors in RA compared with HD. *p<0.05, **p<0.01, ***p<0.001.
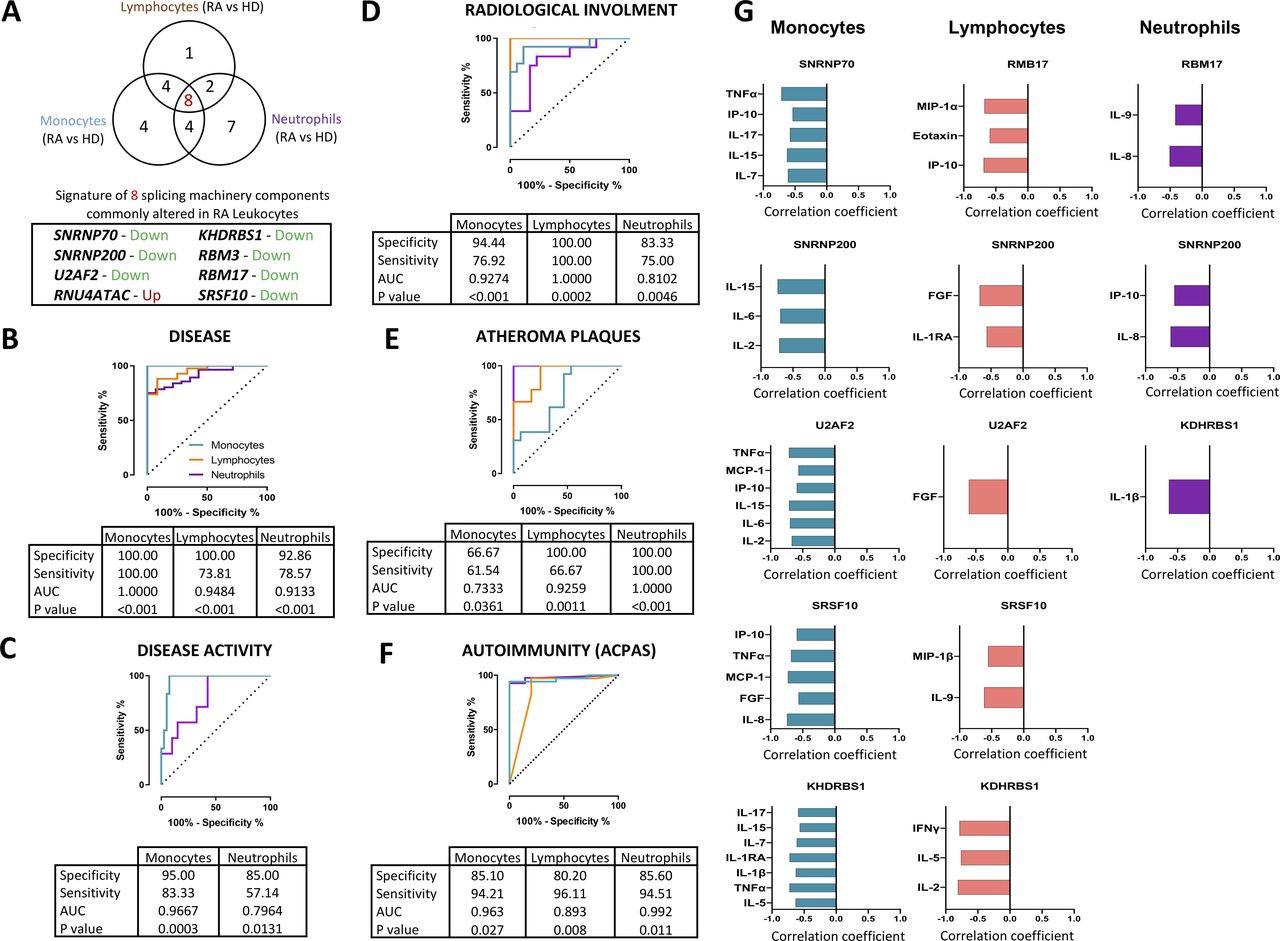
Eight components of the SM, simultaneously altered in the three RA leucocyte subtypes, are related to key clinical features
Eight SM components were found simultaneously altered in the three leucocyte subtypes. These components included major (SNRNP70, SNRNP200 and U2AF2) and minor (RNU4ATAC) spliceosome components and four splicing factors (RBM3, RBM17, KHDRBS1 and SRSF10). All of them were significantly reduced in the three leucocyte subsets of patients with RA, except for RNU4ATAC, which was consistently overexpressed (figures 1 and 2A). Of note, a significant relationship among all those components was identified (online supplemental figure 1).

A signature of eight components of the splicing machinery is commonly altered in RA leucocytes and associated with clinical features of RA. (A) Venn diagram of differentially expressed splicing machinery elements in RA versus HD in leucocyte subtypes (monocytes, lymphocytes and neutrophils). A signature of eight spliceosome components commonly altered in all cell types are also highlighted indicating the direction of that alteration. The potential of this signature in each cell type as biomarkers of disease (B), disease activity (C), radiological involvement (D), atheroma plaques (E) and ACPAs positivity (F) were further demonstrated through logistic regression and receiver operating characteristic curve analysis. Area under the curve (AUC), specificity, sensitivity and p value are displayed in each analysis. (G) Correlation analysis between the signature of eight spliceosome components in each leucocyte subtype and the plasma pro-inflammatory profile was performed, and those showing a p<0.05 are shown. Spearman correlation coefficient is displayed where appropriate. ACPAs, anti-citrullinated protein antibodies; FG, fibroblast growth factor; HD, healthy donors; IFN, interferon; IL, interleukin; IP, interferon gamma-induced protein; MCP, monocyte chemoattractant protein; RA, rheumatoid arthritis; TNF, tumour necrosis factor.
Consequently, we next sought to ascertain if these eight components might be used as potential biomarkers of disease. Thus, we developed different mathematical models by applying logistic regressions on the data sets.
First, we generated a model that clearly discriminated between patients with RA and HD, with area under the curve always above 0.9 and with high specificity (figure 2B). Then, we created models to categorise different RA subsets, allowing to classify: (1) high disease-activity patients—that is, those presenting a DAS28-score higher than 5.1—(figure 2C); (2) patients suffering radiological involvement (figure 2D); (3) patients exhibiting atheroma plaques identified by doppler ultrasonography (figure 2E); and (4) patients positive for ACPAs versus those negative for these autoantibodies (figure 2F). All these models showed high specificity, particularly those generated in monocytes and lymphocytes.
The eight components of the SM, simultaneously altered in the three RA leucocyte subtypes, are closely related to their inflammatory profile
Bio-Plex analyses recognised an inflammatory profile in plasma of patients with RA, on which patients displayed altered expression of several ILs, chemokines and growth factors involved in inflammation and migration to inflamed tissues (online supplemental figure 2). Interestingly, a number of those inflammatory mediators were closely linked to the altered expression of the eight common components, although in a specific way on each leucocyte subtype (figure 2G).
The SM was deeply altered in the joints of both patients with RA and a RA mouse model
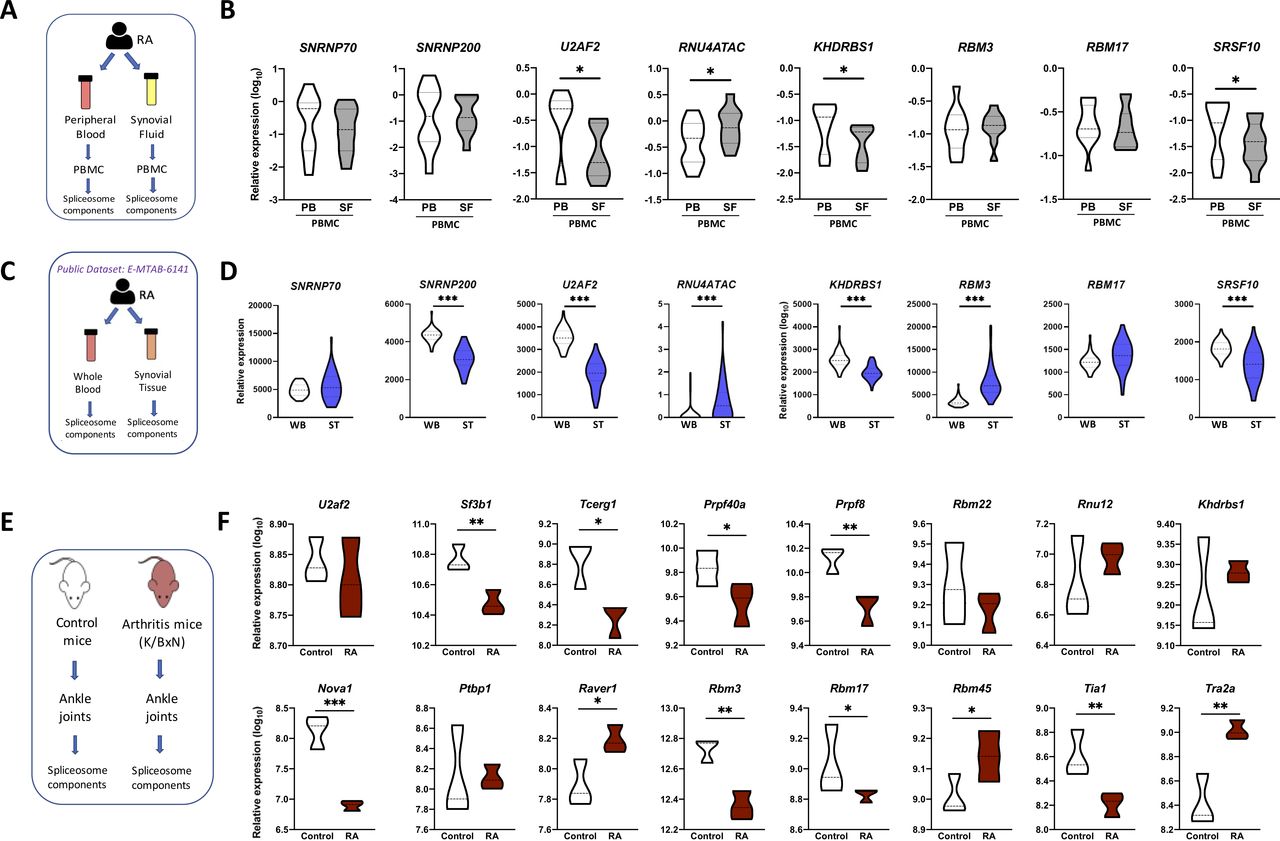
To reinforce the biological relevance of our findings, we evaluated those eight common components in mononuclear cells isolated from the synovial fluid of 15 patients with RA and compared their expression with that of mononuclear cells from peripheral blood of the same patients with RA. Four out of the eight common components were even more altered in synovial fluid mononuclear cells (figure 3A,B), so that U2AF2, KHDRBS1 and SRSF10 were significantly lower in mononuclear cells from synovial fluid than from peripheral blood and, consistently, RNU4ATAC was even higher.

The splicing machinery is deeply altered in the joints of both patients with RA and RA mouse model. (A) Schematic representation of the analysis of the spliceosome signature in paired samples of RA PBMC from peripheral blood and synovial fluid. (B) Violin plots representing the expression levels of the spliceosome signature in PBMC of 15 patients with RA. (C) Schematic representation of the analysis of spliceosome signature in paired samples of whole blood and synovial tissue from patients with RA using public RNA-seq data set (E-MTAB-6141). (D) Violin plots representing the expression levels of the spliceosome signature in 44 patients with RA. (E) Schematic representation of the analysis of the spliceosome machinery in joints from K/BxN arthritis mouse model. (F) Violin plots representing the expression levels of the mouse spliceosome machinery components. *p<0.05, **p<0.01. HD, healthy donors; PB, peripheral blood; PBMC, peripheral blood mononuclear cells; RA, rheumatoid arthritis; SF, synovial fluid; ST, synovial tissue; WB, whole blood.
These results were further validated externally using an independent public RNA-seq data set (E-MTAB-6141). A significant match with our results regarding the expression of the eight-SME signature was demonstrated. Thus, U2AF2, RNU4ATAC, KHDRBS1 and SRSF10 expression were altered in the same fashion, while downregulation and upregulation of SNRNP200 and RBM3, respectively, were observed in this cohort (figure 3C,D). Likewise, the analysis of the 37 remaining SME evaluated in our study in this new cohort showed that a significant proportion of them displayed a lower expression in synovial tissue samples when compared with whole blood (online supplemental figure 3).
In line with this, the expression of SM components in ankle joints of the K/BxN-arthritic mice showed a marked dysregulation compared with control mice. Thus, 11 out of the 16 genes belonging to the SM assessed in the gene array were found altered in the ankles of K/BxN-arthritic mice (figure 3E,F). Hence, although arthritic mice did not fully mimic that found in humans, they display parallel alterations on the SM. Moreover, some components are altered in the same way in both, human and animal models.
The SME expression pattern is associated with differential splice events, splice variants and gene pathways
The public RNA-seq data set of patients with RA was divided according to the high or low expression of each altered SME. First, we analysed the differential generation of splicing variants. Next, we evaluated the impact of differential SME levels in the global performance of the splicing process. Finally, we explored enriched gene pathways associated with the differential SME expression.
The unsupervised analysis of splicing variants revealed the presence of distinctive levels of specific isoforms when comparing high or low expression of the studied components (online supplemental figure 4A). Interestingly, the differential expression profiles of the splicing variants were specific to each of the SME analysed.
RBM17 comprise the highest number of differentially expressed splicing variants (104), while SNRNP200 showed the lowest number of them (21) (online supplemental figure 4C). It should be also noted that three splicing variants were differentially expressed among all the analysed elements. Remarkably, we observed that two out of these three splicing variants belonged to the ITGA11 gene, an alpha integrin that acts as a collagen receptor, playing a potential role in RA development.
Besides, when comparing high and low expression of selected components, we observed a global alteration in the generation of alternative splicing events. Specifically, retained intron, skipped exon and mutually exclusive exons events occurred differentially. Particularly, in the case of U2AF2—a member of the core of the spliceosome—those samples presenting lower U2AF2 levels displayed: (1) more exon skipping, (2) alternative 3’-splicing and 5’-splicing site, (3) less mutually exclusive exon and (4) alternative first and last exons, than those samples presenting high U2AF2 levels (online supplemental figure 4B).
Additionally, to elucidate the particular pathways potentially affected by the dysregulation of these SME, we assessed enrichment analyses on both, differentially spliced variants and differentially expressed genes associated to the studied SME.
Concerning differentially expressed spliced variants, pathways related to RA pathogenesis, such as ‘immune response’, ‘response to TNF’, ‘interferon signalling’ and ‘toll-like receptor signalling’ were frequently observed (online supplemental figure 5). Similarly, among the enriched pathways according to differentially expressed genes, several pathways involved in immune function and inflammation, such as ‘T-helper immune response’, ‘interferon signalling’, ‘interleukin-1 beta biosynthesis’ or ‘interleukin-2 production’ were noticed (online supplemental figure 6).
Anti-TNF therapy modified expression of altered SM genes in RA leucocytes
Within the cohort of patients with RA treated with TNFi, according to DAS28-response,23 66% were responders, while 34% were non-responders (figure 4A). TNFi therapy for 3 months reverted the alteration observed in four commonly dysregulated SME in lymphocytes from responder patients (SNRNP200, U2AF2, KHDRBS1 and RBM17) (figure 4B), promoting a significant upregulation, while in non-responders no changes were observed. No effects were observed in monocytes or neutrophils (data not shown).

Anti-TNF therapy reverse the altered spliceosome signature of lymphocytes along with the inflammatory and clinical profile of patients with RA. (A) Table showing clinical and serological characteristics of 38 patients with RA before and after 3 months of TNFi therapy. Data are divided in responders and non-responders based on European League Against Rheumatism guidelines. (B) Heat map showing levels of circulating inflammatory molecules in plasma of patients with RA before and after 3 months of TNFi therapy. Levels of inflammatory molecules are expressed as log 2 and normalised to time 0 (T0), before therapy, in responders and non-responders’ patients with RA. (C) Violin plots representing the expression distribution of the eight spliceosome components in lymphocytes before and after 3 months of TNFi therapy in responders and non-responders’ patients with RA. *p<0.05, **p<0.01. ACPAs, anti-citrullinated protein antibodies; CRP, C-reactive protein; DAS28, disease activity score-28; ESR, erythrocyte sedimentation rate; FGF, fibroblast growth factor; HAQ, health assessment questionnaire; IL, interleukin; IP, interferon gamma-induced protein; MCP, monocyte chemoattractant protein; MIP, macrophage inflammatory protein; R, responders, patients with RA; NR, non-responders, patients with RA; TNF, tumour necrosis factor; TNFi, TNF inhibitor; T0, time before TNFi therapy; T3, time 3 months after TNFi therapy; VAS, visual analogue scale; VEGF, vascular endothelial growth factor; Y, years.
As previously reported,36 these alterations in responder’s patients with RA paralleled the downregulation of key inflammatory mediators in plasma such as FGF, IL-2, IL-6, IL-8, TNFα and IP-10, while no changes were observed in non-responders patients with RA (figure 4C). Changes in SME did not correlate with those of ACPAs, which did not significantly change after TNFi therapy.
To further confirm the novel role of TNFi therapy as modulator of SME expression, we analysed the changes promoted by a longer period of TNFi treatment in whole blood samples from responders’ patients of the same RA cohort. These analyses showed the reversion in five SME (SNRNP200, KHDRBS1, RBM17, SNRNP70 and SRSF10) after 6 months of TNFi therapy. Accordingly, along with the clinical disease improvement, a simultaneous downregulation of the same inflammatory mediators in plasma was confirmed in these patients after 6 months of treatment (online supplemental figure 7).
Lastly, statistical analyses did not confirm a potential role of SME levels as predictors of TNFi response (data not shown).
In vitro stimulation of healthy leucocytes with ACPAs mimicked the alteration of the SM trough FcR-dependent mechanisms
Given the key role of ACPAs in the pathophysiology of RA,37 we wondered if they could have a role in the dysregulated expression of the SM components observed in vivo. Indeed, in vitro treatment of HD peripheral blood leucocytes with purified ACPAs obtained from RA serum through CCP-affinity columns, clearly altered the expression of the eight commonly altered components, although in a specific way in each leucocyte subtype, being the changes promoted in lymphocytes and monocytes the most relevant (figure 5A–C). Likewise, the inflammatory profile of leucocytes was upregulated (figure 5D–F).

In vitro treatment of healthy leucocytes with ACPAs modify the expression of the spliceosome signature along with their associated inflammatory profile. Monocytes (A,D), lymphocytes (B,E) and neutrophils (C,F) from healthy donors were treated with 10 μg/mL of either IgG-ACPA purified from patients with RA through CCP-affinity column chromatography (IgG-ACPAs(+)) or the flow through depleted in Ig-ACPAs (IgG-ACPAs(-)) for 24 in monocytes and lymphocytes and 6 hours in neutrophils. Spliceosome components (A,B,C) and inflammatory molecules (D,E,F) were analysed by RT-PCR. Data from five independent experiments carried out in triplicate are shown. *p<0.05, **p<0.01. ACPAs, anti-citrullinated protein antibodies; IL, interleukin; TNF, tumour necrosis factor.
To gain insight in the mechanisms related to the modulation of SME in leucocyte subsets by ACPAs, we performed in vitro studies involving blocking the FcR. The downregulation of several SME induced by ACPAs and the parallel upregulation of several inflammatory mediators was prevented by the blockage of FcR (online supplemental figure 8), thus suggesting that the effects of these autoantibodies are mediated, at least partially, by FcR-dependent mechanisms.
To further confirm this specific alteration promoted by ACPAs, we run in parallel another set of experiments where HD leucocytes were treated with monoclonal ACPAs, and IgG control (online supplemental figure 9). Similar results were obtained using this experimental approach, thus supporting the key role of ACPAs in the SME alteration and inflammation.
Next, to explore the potential role of citrullination in the dysregulation of the SM, we evaluated by LC-MS/MS the citrullinome in PBMCs from two sets of patients with RA, including those with severe SME alteration (lower levels) and those with mild SME alteration (higher levels) (online supplemental figure 10A,B).
By this approach, a total of 233 citrullinated peptides in RA were recognised (online supplemental table 2). Patients with severe SME alterations displayed higher degree of global citrullination than those with mild altered SM (online supplemental figure 10C).
Moreover, we further identified a higher degree of citrullination in three of the main established autoantigens in RA: collagen, vimentin and alpha enolase38 39 in patients with RA with severely altered SME compared with patients with RA with mild altered SME (online supplemental figure 10D).
In vitro treatment with inflammatory cytokines dysregulated the SM in HD leucocytes
To evaluate the potential influence of inflammation on the SME dysregulation observed in patients with RA, leucocyte subsets were treated with key cytokines involved in the immune-mediated pathogenesis of RA. TNFa, IL-6 and CCL2 promoted a significant dysregulation in the eight commonly altered SM components, specific for each leucocyte subset, pointing at a relevant role of inflammatory mediators in the control of SM (online supplemental figure 11).
Overexpression of KHDRBS1 and SNRNP70 promoted the downregulation of key inflammatory mediators and functional endpoints in RA
Next, we aimed to prove whether restoration of altered levels of SM factors might have a positive impact in the altered activity of RA leucocytes. Specifically, we evaluated the effects of KHDRBS1 and SNRNP70 overexpression (figure 6A). These genes were selected based on their low expression in the three leucocyte subsets and their clinicals’ associations. The overexpression of these genes reverted inflammatory features to normal-like levels. Specifically, lymphocytes showed a reduction of 10 and 18 inflammatory mediators after KHDRBS1 and SNRNP70 overexpression, respectively, compared with mock transfected cells (figure 6B). Patients with RA-derived monocytes showed reduced cell adhesion after SNRNP70 transfection, (figure 6C). Lastly, neutrophils displayed a downregulation in NETosis features, involving reduced cell-free nucleosomes in response to the overexpression of both genes and a reduction of elastase after SNRNP70 overexpression (figure 6D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In vitro, the induced overexpression of KHDRBS1 and SNRNP70 promote the downregulation of key inflammatory mediators and functional endpoints in RA. Monocytes, lymphocytes and neutrophils from patients with RA were transiently transfected using KHDRBS1 and SNRNP70 plasmid and empty vector (mock) used as control. (A) mRNA expression of KHDRBS1 and SNRNP70 after transfection by RT-PCR. (B) Protein expression of KHDRBS1 and SNRNP70 after transfection by Western Blot analysis. (C) Pro-inflammatory molecules were analysed in lymphocytes’ supernatant using a multiplex assay 24 hours after transfection. Cell adhesion (D) was assessed on monocytes 24 hours after transfection. Cell-free nucleosomes and elastase (E) were evaluated in neutrophils’ supernatant after 6 hours. All experiments were compared with mock transfected cells, which was used as control and set at 100% in each panel. *p<0.05, **p<0.01, ***p<0.001. FGF, fibroblast growth factor; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IFN, interferon; IL, interleukin; IP, interferon gamma-induced protein; MCP, monocyte chemoattractant protein; MIP, macrophage inflammatory protein; mRNA, messenger RNA; PDGF, platelet derived growth factor; RA, rheumatoid arthritis; TNF, tumour necrosis factor; VEGF, vascular endothelial growth factor.
Modulation of the leucocyte-SME impact the global function of RA SF
Lastly, we evaluated the potential impact of modulating dysregulated leucocyte-SME in the function of RA SF. The supernatants generated by the induced overexpression of KHDRB1 and SNRNP70 in lymphocytes from patients with active RA ameliorated, in vitro, the aberrant activation status of RA SF through: (1) the reduction of cell migration capacity (online supplemental figure 12B); (2) the decrease of the proliferation rate (online supplemental figure 12C); and (3) the downregulation in the levels of both inflammatory mediators and extracellular matrix components (online supplemental figure 12D).
Discussion
The present study demonstrates, for the first time, that the SM is profoundly altered in RA leucocytes and closely linked to the pathophysiology of the disease. Importantly, we identified eight components commonly altered in leucocytes subsets, whose expression levels enabled distinguishing patients with RA from HD, and identifying patients with high disease activity, articular involvement and early atherosclerosis. Moreover, we extended these observations by examining the relationship among altered levels of SM components and those of inflammatory mediators notably involved in the clinical profile of these patients. These results were further validated in mononuclear cells obtained from synovial fluid of patients with RA, where inflammatory damage is more pronounced, and on the articular joints of a mouse model of RA, thus reinforcing the clinical relevance of the data obtained. Ex vivo and in vitro studies further identified potential mechanisms underlying these processes. Finally, significant effects of in vivo TNFi therapy on the reversion of SME dysregulation was demonstrated.
We have previously reported that the SM is altered in tumorous, metabolic and chronic inflammatory diseases.24–28 However, this is the first study focused on the analysis of these alterations in patients with RA and their influence in its pathophysiology. The eight elements of the SM found commonly altered in the three leucocyte subsets evaluated included several molecules belonging to the major and minor spliceosome and four splicing factors. All of them are functionally interrelated, but no coordinated alterations had been reported hitherto in the setting of RA.
Several studies have established relevant (dys)functions of some of these factors in leucocytes, including an aberrant expression of SNRNP200 (an essential component of the U5 spliceosome complex) in the cell membrane of leukaemic blasts,40 and an activating role of U2AF2 in CD4+ T cells,.41 RBM3 has shown roles in erythropoietic differentiation and in immune response, inducing the overexpression of cytokines such as IL-6 or TNF α in infection and non-infection conditions.42 Similarly, RBM17 has been identified as modulator of apoptosis, proliferation and cell adhesion.43 Also, SRSF10 has been established as a key modulator of metabolic pathways critical for obesity and related metabolic phenotypes, including adipocyte differentiation44 and atherosclerosis development,45 all of them closely related to the establishment of a chronic inflammatory status. In contrast, the role of RNU4ATAC has been scarcely explored in disease, having been only reported mutations in developmental rare diseases.46–48
The role of SNRNP70 in leucocytes activity has been only superficially explored to date. It has been reported the presence of a protein codified by SNRNP70 (U1-70K autoantigen-specific) in T cells of mixed connective tissue disease (MCTD), which may be used for its diagnosis, distinguishing MCTD from systemic lupus erythematosus.49However, its potential role in RA has not been fully explored. Lastly, KHDRBS1 is overexpressed in fibroblast-like synoviocytes of patients with RA, wherein it was involved in invasion, migration and proliferation.50 Conversely, our RA cohort showed a lower expression of KHDRBS1 in the three leucocyte subsets, accompanied by an even lower expression in synovial fluid leucocytes. Nevertheless, in our cohort, an inverse relationship among reduced levels of this splicing factor and both, a higher disease activity and the overexpression of a number of circulating inflammatory mediators was demonstrated, thus supporting its potential involvement in the pathophysiology of RA and pointing to a specific dysregulation in different cells and tissues.
To improve the significance of our results, we used a public available RNA-seq data set where matching whole blood cells and synovial biopsies of patients with RA were analysed. Even being slightly different samples, involving not only immune cells but the whole blood cell population and synovial tissue, we observed a clear correspondence with our results regarding both, the eight SME signature identified and even the whole set of SME evaluated. Thus, our results, in conjunction with the new evaluated database, support the presence of wide alterations of several SME in immune cells and synovial tissue, involving even more dysregulation in the last one. This prominent alteration in the synovium might be associated to the enhanced local inflammation widely reported in the RA joints.
Next, we analysed the biological consequences of the SME differential expression. These analyses identified, first, a distinctive alteration on the generation of alternative splicing events among patients displaying high or low SME levels. Besides, we recognised differentially expressed splicing variants among patients with RA with high or low SME expression levels, further showing a high specificity linked to each SME. Moreover, enrichment analyses on both, differentially spliced variants and differentially expressed genes associated to the studied SME, identified relevant pathways involved in RA pathogenesis.
That overall data strongly supports the notion that the dysregulation of the studied SME might have a deep impact in the splicing process and the generation of splicing variants and, consequently, might play a key role on the progression of this autoimmune disorder.
Increased protein citrullination is a hallmark of RA, closely associated with the generation of ACPAs. Interestingly, intracellular citrullinated proteins are involved in RNA splicing. In fact, some of them act as components of the RNA SM—including several heterogeneous nuclear ribonucleoproteins and SNRNP200, an essential component of the U5 spliceosome complex51—implying that citrullination modulates RNA biology. In the present study, the analysis of the citrullinome in RA PBMCs by LC-MS/MS identified a number of citrullinated peptides, which showed, a higher degree of citrullination in patients with more severe SM alteration. Moreover, we identified three well-established autoantigens in RA (alpha enolase, vimentin and collagen),38 39 as highly citrullinated in patients with severely altered SM. These results suggest a clear relationship between this post-translational protein modification and the dysregulation of SM in the immune cells of patients with RA.
In addition, the involvement of citrullination in the dysregulation of the SM in RA was in line with both, the relationship among this alteration and ACPA positivity in leucocytes in patients with RA leukocytes, and the in vitro effects of ACPA on SME expression levels, thus suggesting a pivotal role for these autoantibodies in the identified SME alteration in RA. ACPAs have been closely related to joint damage, inflammation, oxidative stress and atherosclerosis in RA, and we previously reported that purified polyclonal ACPAs induced the expression of pro-inflammatory cytokines.52 Accordingly, in vitro treatment of HD leucocytes with ACPAs upregulated a number of cytokines, chemokines and growth factors in distinct immune cells. Notably, in vivo, circulating levels of these pro-inflammatory proteins inversely correlated with the reduced levels of several SM components in leucocytes, pointing at a complex regulatory role of ACPAs in these processes.
In the search for a potential mechanism underlying the SME modulation by ACPAs, we evaluated the potential involvement of FcR, previously reported to mediate the effects of these autoantibodies in leucocyte activation.53 The blockage of FcR prevented the downregulation of SME induced by ACPAs, underlying the role of these receptors on such effects.
In line with this, we also evaluated the influence of key cytokines linked to the pathophysiology of RA—widely reported to be both, associated in vivo to the positivity for ACPAs, and induced in vitro after treatment with these autoantibodies52—in the expression of the dysregulated SME. The in vitro treatment of HD leucocytes with either, TNF, IL-6 or CCL2, promoted a significant dysregulation in the eight commonly altered SME. That data suggested that both, ACPAs and inflammation might contribute either jointly or independently to the SME dysregulation associated to the pathogenesis of this chronic disorder.
Lastly, we evaluated the potential involvement of some dysregulated SME in the pathogenic activity of RA leucocytes. The overexpression of KHDRBS1 and SNRNP70 in monocytes, lymphocytes and neutrophils promoted a downregulation of several inflammatory proteins secreted by lymphocytes, decreased cell adhesion in monocytes and reduced NETosis in neutrophils. These effects were acuter after overexpression of SNRNP70, probably due to its central function in the core of the spliceosome, as responsible for most of splicing processes, while the functional role of KHDRBS1 may be constrained to the splicing of certain genes.
Moreover, in our hands, the modulation of these SME in lymphocytes positively stuck the aberrant activation status of RA SF, reducing their inflammatory profile, along with their proliferative and migration capacities. Overall, these results demonstrated for the first time that the modulation of the SM in leucocytes from patients with RA directly impacts relevant pathogenic functions associated with the disease. Thus, the pharmacological intervention of these components might have a therapeutic potential role in patients suffering this and other immune-mediated diseases.
Anti-TNF therapy has significantly improved the outlook for patients suffering from RA.36 With that premise, we evaluated in a new cohort of patients with RA the in vivo effects of TNFi on the altered expression of SME. Interestingly, in parallel to the early (3 months) and established (6 months) reduction of the disease activity and the efficient downregulation of their inflammatory profile, TNFi significantly reversed the levels of the SME altered in peripheral blood leucocytes. The presence of isoforms of the soluble TNF receptor 2, produced by alternative splicing in RA, has been demonstrated to maintain a prolonged therapeutic response to TNF.17 Overall, although a role of SME as biomarkers for predicting or monitoring therapeutic response was not confirmed, our data support that their reversed expression might constitute an additional and/or complementary mechanism underlying the clinical response to TNFi in RA.
This study has some limitations. First, the specificity of the eight SME signature as biomarker of disease in RA was not confirmed by comparison with other chronic or autoimmune diseases. Second, new extensive cohorts of patients with RA should be evaluated to confirm and validate the alterations observed in the splicing machinery, and the effects promoted by TNFi and other biological therapies. Lastly, despite we have provided several mechanistic insights related to the regulation of the SME, the deep understanding of the mechanisms underlying their pathogenic role and modulation in disease context is still to be fully characterised by the scientific community.
Altogether, we have identified a signature composed of eight elements of the SM, simultaneously dysregulated in immune cells and closely related to key clinical features of patients with RA. Each of these components displays widespread effects on the transcription of multiple genes. Thus, most probably their coordinated altered expression, rather than a unique or specific alteration, would be responsible for the development of clinical profiles, and might jointly influence the therapeutic response to TNFi. Overall, our results reveal, for the first time, the involvement of specific SME on the pathogenesis of RA, their relationship with the inflammatory and autoimmunity status of the disease and their modulation by TNFi therapy, which jointly invite to further explore the targeting of altered splicing as a novel source of therapeutic tools in this autoimmune disorder.
Data availability statement
Data are available upon reasonable request. All data relevant to the study are included in the article or uploaded as supplementary information. It would be also available from the corresponding author upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
Ethics approval was obtained from the ethics committee of the Reina Sofia University Hospital (Córdoba, Spain).
Acknowledgments
The authors thank all patients and healthy subjects for participation in the study. We would like to thank the Proteomics Platform of the Institute for Biomedical Research of A Coruña (INIBIC), ProteoRed-ISCIII, A Coruña, Spain, for their excellent technical assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
AI-C, CP-S and AMP-T are joint first authors.
Handling editor Josef S Smolen
Twitter @avpr1b
Correction notice This article has been corrected since it published Online First. The author's name, Justo P Castaño, has been corrected.
Contributors AI-C, AMP‐T, ML‐T, CR-R, SP-A and MdR-M developed the in vivo assays, performed the experiments and solved technical problems; AE-C, ROC, JC, PS and EC‐E followed-up with patients and contributed useful discussion and suggestions; CC, AG and NB developed the mouse models, performed statistical analysis and discussed results; MCA‐A and IAdlR performed some experiments and analysed the data; AI-C and RB-E performed bioinformatic and biostatistical analyses. AI-C, CP‐S, RML, JPC-F and CL‐P formed the hypothesis, directed and coordinated the project, designed the experiments, analysed the data and wrote the manuscript. EC-E and CL-P are shared last authorship. EC-E is also a senior contributor.
Funding This study was supported by grants from the Instituto de Salud Carlos III (PI18/00837), cofinanciado por el Fondo Europeo de Desarrollo Regional de la Unión Europea ‘Una manera de hacer Europa’, Spain, the Andalusian Regional Health System (ref. PI‐0285‐2017) and the Spanish Inflammatory and Rheumatic Diseases Network (RIER), Instituto de Salud Carlos III (RD16/0012/0015). CL‐P was supported by a contract from the Spanish Junta de Andalucía (‘Nicolas Monardes’ programme). AI-C was supported by ‘Juan de la Cierva’ and ‘Sara Borrell’ programmes (FJCI‐2016‐30825 and CD19/00255).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.