Article Text

Abstract
Objectives To evaluate efficacy and safety of the anti-interleukin-23p19 monoclonal antibody tildrakizumab in patients with psoriatic arthritis (PsA).
Methods In this randomised, double-blind, placebo-controlled, phase IIb study, patients with active PsA were randomised 1:1:1:1:1 to tildrakizumab 200 mg every 4 weeks (Q4W); tildrakizumab 200, 100 or 20 mg Q12W; or placebo Q4W. Patients receiving tildrakizumab 20 mg or placebo switched to tildrakizumab 200 mg Q12W at W24; treatment continued to W52. The primary efficacy endpoint was proportion of patients with ACR20 response (≥20% improvement by American College of Rheumatology criteria) at W24. Secondary efficacy endpoints were assessed without adjustment for multiplicity. Safety was evaluated from treatment-emergent adverse events (TEAEs).
Results 391/500 patients screened were randomised and treated. At W24, 71.4%–79.5% of tildrakizumab-treated versus 50.6% of placebo-treated patients achieved ACR20 (all p<0.01). Patients receiving tildrakizumab versus placebo generally achieved higher rates of ACR50, Disease Activity Score in 28 joints with C reactive protein <3.2, minimal disease activity and 75%/90%/100% improvement from baseline Psoriasis Area and Severity Index responses at W24 and through W52. Improvement in dactylitis and enthesitis was not observed; results were mixed for other outcomes. Responses in patients switched to tildrakizumab 200 mg at W24 were consistent with treatment from baseline. TEAEs and serious TEAEs occurred in 64.5% and 3.3%, respectively, of all patients through W52 and were comparable among treatment arms.
Conclusions Tildrakizumab treatment significantly improved joint and skin manifestations of PsA other than dactylitis and enthesitis. Treatment was generally well tolerated through W52. Clinicaltrials.gov NCT02980692.
- arthritis
- psoriatic
- biological therapy
- cytokines
Data availability statement
Data are available upon reasonable request. Data and other documents will be made available after publication, with no end date, to anyone who submits a reasonable request to the study sponsor.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
There is an unmet need for psoriatic arthritis (PsA) therapies that maximally address all clinical manifestations of the disease and improve patient quality of life.
Interleukin (IL)-23 is a key regulatory cytokine in the pathogenesis of PsA, and the p19 subunit of IL-23 is an effective therapeutic target for PsA in clinical studies; tildrakizumab is a high-affinity anti-IL-23p19 monoclonal antibody approved in the USA, Europe, Australia and Japan for treatment of plaque psoriasis.
What does this study add?
This study demonstrates that tildrakizumab was superior to placebo in achieving ACR20 (≥20% improvement by American College of Rheumatology criteria) and ACR50 responses, minimal disease activity; Disease Activity Score in 28 joints with C reactive protein <3.2 and ≥75% improvement from baseline Psoriasis Area and Severity Index (PASI 75), PASI 90 and PASI 100 responses at week 24; response rates were sustained through week 52. Improvement in dactylitis and enthesitis was not observed, and results for other outcomes were mixed among patients receiving different doses of tildrakizumab.
Tildrakizumab was generally well tolerated with no reports of uveitis, systemic fungal infections, inflammatory bowel disease, major adverse cardiac events or deaths through week 52.
How might this impact on clinical practice or future developments?
These findings support the efficacy and safety of tildrakizumab in patients with PsA and the planned dosing schedules in the ongoing phase III clinical programme.
Introduction
Psoriatic arthritis (PsA) is a chronic, progressive, inflammatory arthritis with estimated global prevalence of 0.2%–0.3%.1–4 Manifestations of PsA include musculoskeletal and skin disease activity; pain; fatigue; systemic inflammation and their effects on physical function, activities of daily living and health-related quality of life (QoL).3 5 6 Chronic joint inflammation and potential joint damage from PsA can impose considerable economic burden.5 There is an unmet need for therapies that address all clinical manifestations of PsA and improve patient QoL.
Treatments for PsA include non-pharmacological therapies, non-steroidal anti-inflammatory drugs, conventional systemic disease modifying antirheumatic drugs (csDMARDs; including methotrexate, sulfasalazine and leflunomide), biological DMARDs (bDMARDs) and targeted synthetic DMARDs (including Janus-associated kinase and phosphodiesterase inhibitors).7–9 Treatment guidelines recommend csDMARDs before other therapies,8 bDMARDs targeting tumour necrosis factor α (TNFα) before csDMARDs,9 or either approach.7
Interleukin (IL)-23 is a key regulatory cytokine in PsA pathogenesis.10 11 Targeting the IL-23/IL-12 p40 subunit with ustekinumab was effective and generally well tolerated in PsA clinical trials.12 13 The anti-IL-23p19 subunit antibody guselkumab, which targets IL-23 alone, was also effective and is approved in the USA for treatment of signs and symptoms of PsA.14–17 Neither agent provided incremental improvement over TNFα inhibitors. Tildrakizumab, a high-affinity anti-IL-23p19 monoclonal antibody, is approved in the USA, Europe, Australia and Japan for treatment of plaque psoriasis.18–24 This phase IIb study evaluated tildrakizumab efficacy and safety in patients with PsA at week 24 and through week 52 (clinicaltrials.gov NCT02980692).
Methods
Study design
This phase IIb, randomised, double-blind, multidose, placebo-controlled, multicentre study was conducted at 74 sites (including hospital dermatology units, specialty clinics, private practices and research sites) in 8 countries. All patients provided written informed consent. In part 1 (weeks 0–24), patients were randomised 1:1:1:1:1 to receive subcutaneous tildrakizumab 200 mg every 4 weeks (Q4W); tildrakizumab 200, 100 or 20 mg every 12 weeks (Q12W) or placebo Q4W (online supplemental figure S1). At week 24, patients receiving tildrakizumab 20 mg or placebo switched to tildrakizumab 200 mg Q12W. All treatments continued in part 2 (weeks 25–52, double-blind follow-up), followed by a 20-week washout period (to week 72) or rollover to the long-term extension (clinicaltrials.gov NCT03552276). This publication reports efficacy and safety outcomes for patients on treatment (through week 52).
Supplemental material
All patients received study drug or placebo Q4W to maintain the blind through week 52; placebo was administered between tildrakizumab doses for patients receiving Q12W dosing. Randomised patients were stratified by prior anti-TNFα therapy use (yes/no; prior anti-TNFα use capped at 30% of total patients) and baseline body weight (≤90 kg/>90 kg). Randomisation was computer generated before the study; patients were allocated to treatment arms using an interactive voice recognition service (ICON Clinical Research, Dublin, Ireland). Patients without minimal response to treatment (<10% improvement from baseline swollen joint count in 66 joints (SJC66) and tender joint count in 68 joints (TJC68)) at week 16 could adjust background medications per maximum permitted dosing.
Patients
Eligible patients were ≥18 years old, with a diagnosis of PsA by the Classification Criteria for Psoriatic Arthritis for ≥6 months25 and had TJC68 ≥3 and SJC66 ≥3 according to an independent assessor. Allowed and prohibited medications are described in online supplemental methods.
Efficacy assessments
The primary efficacy endpoint was proportion of patients with an ACR20 response (≥20% improvement by American College of Rheumatology criteria) at week 24. Prespecified secondary endpoints included proportions of patients achieving ACR20 at week 52 and ACR50, ACR70, Disease Activity Score in 28 joints with C reactive protein (DAS28-CRP) <3.2 and minimal disease activity (MDA) at weeks 24 and 52 or requiring background medication adjustment at week 24; and change from baseline in individual ACR components, Leeds Dactylitis Index (LDI; in patients with baseline LDI ≥1), Leeds Enthesitis Index (LEI; in patients with baseline LEI ≥1) and Health Assessment Questionnaire-Disability Index (HAQ-DI) at weeks 24 and 52. Patients achieved MDA if they met 5 of 7 criteria—TJC68 ≤1, SJC66 ≤1, Psoriasis Area and Severity Index (PASI) ≤1 or body surface area (BSA) ≤3%, patient pain visual analogue scale (VAS) ≤15, patient global disease activity VAS ≤20, HAQ-DI ≤0.5 and tender entheseal points ≤1. Proportions of patients achieving 75%/90%/100% improvement from baseline PASI (PASI 75/90/100) for patients with measurable psoriasis (baseline affected BSA ≥3%) and PsA Impact of Disease (PsAID)26 change from baseline at weeks 24 and 52 were exploratory efficacy endpoints. Post hoc analyses (online supplemental methods) included proportions of patients achieving very low disease activity (VLDA), Psoriatic Arthritis Disease Activity Score (PASDAS) <3.2, Disease Activity in Psoriatic Arthritis (DAPSA) remission (score 0–4), complete LDI/LEI resolution and minimum clinically important difference (MCID) change from baseline HAQ-DI (≥0.35) and PsAID (≥3) at weeks 24 and 52; DAPSA/PASDAS change from baseline; and median LDI/LEI.27 The TJC, SJC, LDI/LEI and PASI assessments were performed by an independent assessor. Other assessment details are summarised in online supplemental methods.
Safety assessments
Safety endpoints included treatment-emergent adverse events (TEAEs), serious TEAEs and TEAEs of special and clinical interest. TEAEs were coded by Medical Dictionary of Regulatory Activities V.20.1 and defined as any AE occurring or worsening on/after the day of first dose of study drug up to week 52 or on/before last dosing date if the patient discontinued treatment. TEAEs of special interest were severe infection, malignancy (including non-melanoma and melanoma skin cancer), confirmed major adverse cardiovascular event (MACE) or drug-related hypersensitivity reaction (details in online supplemental methods). TEAEs of clinical interest included any non-serious TEAE considered of special interest and reported to the sponsor similarly to a serious TEAE (details in online supplemental methods). Routine laboratory investigations and physical examinations were performed, and vital signs were monitored at screening and throughout the study. To ensure patient safety, an independent data safety monitoring board routinely reviewed data and provided the sponsor with recommendations.
Statistical analyses
The number of patients enrolled was based on assumed ACR20 response rates of 30%, 35% and 50% for placebo, tildrakizumab 20 mg, and higher doses of tildrakizumab, respectively; a one-sided alpha=0.05; 80.7% power and a 5% dropout rate. All analyses were performed using SAS V.9.4 or later.28 Efficacy and safety analyses included all randomised patients who received ≥1 dose of study drug or placebo (full analysis set). Statistical comparison of ACR20/50/70, PASI 75/90/100 and MDA response rates between tildrakizumab arms versus placebo used the Cochran-Mantel-Haenszel test, stratified by prior anti-TNFα therapy use and baseline body weight. Two-sided 95% CIs and p values were calculated for each tildrakizumab treatment arm versus placebo. Non-response imputation (NRI) was used for patients who withdrew from the study or had incomplete data at week 52 unless otherwise specified. Patients who required adjustments to background medications were counted as non-responders for the primary analysis. Continuous endpoints were analysed by mixed-model repeated measure analysis with fixed effects of treatment, visit, treatment by visit interaction, prior anti-TNFα therapy use (yes/no), baseline bodyweight (≤90 kg/>90 kg) and baseline value; missing data were imputed using NRI. For the primary endpoint of ACR20 response at week 24, Type I error was controlled using the Simes testing procedure (online supplemental methods); there was no multiplicity adjustment for secondary endpoints. No formal hypothesis testing was performed for post hoc analyses.
Patient and public involvement
Patients and the public were not involved in study design, recruitment or dissemination of results, and patients were not asked to assess the burden of study participation.
Results
Patients
From 19 April 2017, to 25 April 2018, 500 patients were screened, of whom 391 were randomised to tildrakizumab 200 mg Q4W (n=78), tildrakizumab 200 mg Q12W (n=79), tildrakizumab 100 mg Q12W (n=77), tildrakizumab 20→200 mg Q12W (n=78) and placebo Q4W→tildrakizumab 200 mg Q12W (n=79) (figure 1). Overall, 331 (84.7%) patients completed part 1 and 315 (80.6%) completed part 2. By week 52, 76 (19.4%) patients discontinued, most commonly due to lack of efficacy (9.5%) or withdrawn consent (3.3%). The last follow-up was on 5 October 2019.

Patient status to week 52. PBO, placebo; Q4W, every 4 weeks; Q12W, every 12 weeks; W, week.
Demographics were comparable between treatment arms (table 1). Of patients analysed, 91 (23.3%) were anti-TNFα therapy-experienced. Across treatment arms, mean duration of PsA was 6–7.5 years, 53%–71% of patients had moderate-to-severe psoriasis (BSA ≥3%) and >60% of patients were receiving csDMARDs with or without corticosteroids at baseline (table 1). Baseline TJC68, patient global assessment (PtGA), physician global assessment (PGA) and patient pain assessment were lower among patients receiving tildrakizumab 200 mg Q4W versus other treatments. Less than half of patients had measurable dactylitis at baseline, and baseline LDI was higher among patients receiving tildrakizumab 100 mg Q12W and placebo Q4W→tildrakizumab 200 mg Q12W, and lower among patients receiving tildrakizumab 200 mg Q4W, compared with other treatments.
Demographics and baseline clinical disease characteristics
Efficacy
At week 24, a significantly higher proportion of patients receiving any dose of tildrakizumab achieved ACR20 (71.4%–79.5%) relative to placebo-treated patients (50.6%) (figure 2A, table 2, all p≤0.0125), with more responders to tildrakizumab 200 mg Q4W and 100 mg Q12W by week 8—after one dose of study medication.

Response rates for (A) ACR20, (B) ACR50 and (C) ACR70 through week 52. Supporting values shown in online supplemental table S3. Missing responses were imputed as non-responses. Shown for randomised patients who received ≥1 dose of study drug. TIL 200 mg Q4W, n=78; TIL 200 mg Q12W, n=79; TIL 100 mg Q12W, n=77; TIL 20 mg Q12W→200 mg Q12W, n=78; PBO Q4W→TIL 200 mg Q12W, n=79. *p<0.05; †p<0.001; ‡p<0.0001 versus PBO; not adjusted for multiplicity, except ACR20 at week 24. P values were not analysed beyond week 24. ACR, American College of Rheumatology; PBO, placebo; Q4W, every 4 weeks; Q12W, every 12 weeks; TIL, tildrakizumab.
Efficacy outcomes at week 24
The secondary endpoints, subgroup analyses and exploratory efficacy endpoints were not multiplicity controlled; nominal p values are provided for information only. At week 24, patients receiving tildrakizumab 200 mg (Q4W or Q12W) achieved higher rates of ACR50/70, DAS28-CRP <3.2 and MDA and greater improvement in PtGA, PGA, patient pain assessment and high-sensitivity CRP (hsCRP) level relative to placebo-treated patients (nominal p<0.05; table 2; figure 2B,C; figure 3A,B; proportions of patients with ≥20% improvement in ACR components over time online supplemental figure S2; change in patient pain assessment over time online supplemental figure S3). Improvement in SJC66 was greater for patients receiving tildrakizumab 200 mg Q4W but not Q12W, and improvements in TJC68 and HAQ-DI were greater for patients receiving tildrakizumab 200 mg Q12W but not Q4W, relative to placebo-treated patients (nominal p<0.05). Patients receiving tildrakizumab 100 mg Q12W versus placebo achieved higher rates or greater improvement in the same outcomes except ACR70; patients receiving tildrakizumab 20 mg Q12W achieved higher rates of ACR50, DAS28-CRP <3.2 and MDA and greater improvement in PtGA, PGA and hsCRP level, but not other measures, relative to placebo-treated patients (nominal p<0.05). Responses were maintained through week 52, and patients who switched from placebo or tildrakizumab 20 mg to tildrakizumab 200 mg Q12W at week 24 had similar responses as patients treated from baseline (figures 2–3, online supplemental table S1, online supplemental figure S3). Improvement in LDI and LEI at week 24 was not observed following any dose of tildrakizumab versus placebo (table 2, online supplemental figure S4). Only two patients required adjustment of background medication (table 2).

MDA responders (A) over time, (B) responders for each MDA subcomponent at week 24 and (C) VLDA responders by treatment and time point. Supporting values shown in online supplemental table S4. Shown for randomised patients who received ≥1 dose of study drug. Error bars represent 95% CI. Missing responses were imputed as non-responses. Proportion of responders shown as % in (B). TIL 200 mg Q4W, n=78; TIL 200 mg Q12W, n=79; TIL 100 mg Q12W, n=77; TIL 20 mg Q12W→200 mg Q12W, n=78; PBO Q4W→TIL 200 mg Q12W, n=79 except for tender entheseal points ≤1 in (B) (TIL 200 mg Q4W, n=76; TIL 100 mg Q12W, n=76; PBO Q4W→TIL 200 mg Q12W, n=78).*p<0.05; †p<0.001; ‡p<0.0001 versus PBO; not adjusted for multiplicity. P values were not analysed beyond week 24. BSA, body surface area; HAQ-DI, Health Assessment Questionnaire-Disability Index; MDA, Minimum Disease Activity; PASI, Psoriasis Area and Severity Index; PBO, placebo; PtGA, patients global assessment; Q4W, every 4 weeks; Q12W, every 12 weeks; TIL, tildrakizumab; VAS, visual analogue scale; VLDA, very low disease activity.
Subgroup analysis by prior anti-TNFα therapy experience was performed for ACR20/50/70 response rates. Week 24 response rates were numerically lower in anti-TNFα-experienced versus anti-TNFα-naïve patients within each treatment arm, but the ACR20/50/70 treatment response pattern was generally similar regardless of prior anti-TNFα therapy (online supplemental figure S5). ACR20 response rates by country are shown in online supplemental table S2; lower proportions of patients receiving tildrakizumab 200 mg or placebo achieved ACR20 response in the USA and Spain relative to other countries.
In exploratory and post hoc analyses, greater proportions of patients with measurable psoriasis at baseline (BSA ≥3%) achieved PASI 75/90/100 at week 24 following treatment with tildrakizumab (any dose) versus placebo, with sustained response through week 52 (figure 4). Impact of PsA on patients’ lives, assessed via PsAID, decreased for patients receiving tildrakizumab (all doses) versus placebo (table 2); improvement was sustained through week 52 (online supplemental table S1). The proportion of patients with VLDA was numerically greater for tildrakizumab 200 mg Q4W and Q12W versus placebo by week 24 (figure 3C, table 2); no hypothesis testing was performed. Tildrakizumab treatment did not increase combined LDI/LEI resolution (table 2) relative to placebo at week 24. At week 52, ≥50% of patients with baseline LEI ≥1 had LEI resolution (online supplemental table S1). DAPSA and PASDAS scores numerically decreased and proportions of patients achieving DAPSA remission and PASDAS <3.2 were numerically larger following treatment with tildrakizumab versus placebo at week 24 and through week 52 (table 2, figure 5, online supplemental table S1); no hypothesis testing was performed. Proportions of patients achieving MCID from baseline HAQ-DI and PsAID at week 24 appeared similar among treatment arms (table 2).
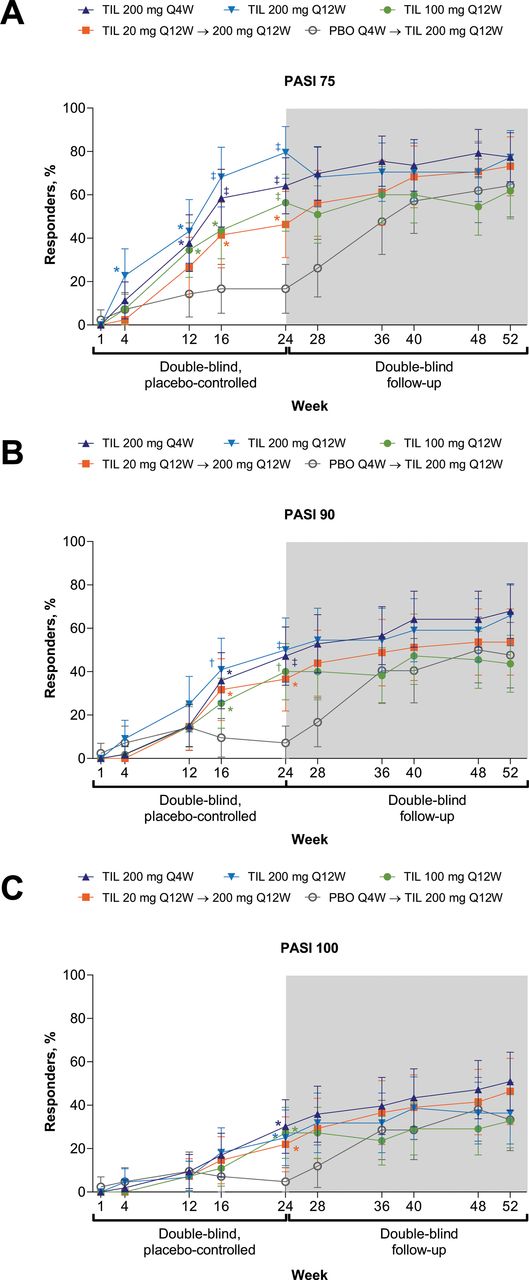
Response rates for (A) PASI 75, (B) PASI 90 and (C) PASI 100 through week 52 across treatment and time point. Supporting values shown in online supplemental table S6. Response rates were calculated in randomised patients who received ≥1 dose of study drug with BSA ≥3% at baseline. Error bars represent 95% CI. Missing responses were imputed as non-responses. TIL 200 mg Q4W, n=53; TIL 200 mg Q12W, n=44; TIL 100 mg Q12W, n=55; TIL 20 mg Q12W→200 mg Q12W, n=41; PBO Q4W→TIL 200 mg Q12W, n=42. P values are based on Cochran-Mantel-Haenszel test (with prior anti-TNF use and baseline weight as stratification factors). *p<0.05; †p<0.001; ‡p<0.0001 versus PBO; not adjusted for multiplicity. P values were not analysed beyond week 24. BSA, body surface area; PASI, Psoriasis Area and Severity Index; PBO, placebo; Q4W, every 4 weeks; Q12W, every 12 weeks; TIL, tildrakizumab; TNF, tumour necrosis factor.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proportion of patients in remission based on (A) DAPSA, proportion of patients with PASDAS <3.2 (B), and change from baseline DAPSA (C) and PASDAS (D). Supporting values shown in online supplemental table S5. Missing responses were imputed as non-responses. DAPSA remission was defined as a score between 0 and 4. TIL 200 mg Q4W, n=78; TIL 200 mg Q12W, n=79; TIL 100 mg Q12W, n=77; TIL 20 mg Q12W→200 mg Q12W, n=78; PBO Q4W→TIL 200 mg Q12W, n=79. P values not analysed. DAPSA, Disease Activity in Psoriatic Arthritis; LS, least squares; PASDAS, Psoriatic Arthritis Disease Activity Score; PBO, placebo; Q4W, every 4 weeks; Q12W, every 12 weeks; TIL, tildrakizumab.
Safety
There were no deaths through study week 52. Of 391 patients analysed, 1 (0.3%) patient discontinued due to a TEAE (hypertension, tildrakizumab 200 mg Q12W). Across all treatment arms, 252 (64.5%) patients had a TEAE, most frequently nasopharyngitis (8.4%) and upper respiratory tract infection (6.4%) (table 3). Two (0.5%) patients had a fungal skin infection (candida, both tildrakizumab 200 mg Q4W). Most TEAEs, including infections, were mild and comparable among treatment arms. During weeks 25–52, one patient (0.3%) was diagnosed with malignancy (intraductal proliferative breast lesion, tildrakizumab 20→200 mg Q12W).
Summary of safety findings
Serious TEAEs were observed in nine (3.3%) patients. One serious infection (chronic tonsillitis) was reported during the first 24 weeks (tildrakizumab 20 mg Q12W). One case each of pyelonephritis and urinary tract infection were reported as TEAEs of special interest in the same patient (tildrakizumab 100 mg Q12W) (table 3). There were no reports of systemic candidiasis, uveitis, inflammatory bowel disease, MACE, suicidality or deaths, and no changes in laboratory parameters considered serious TEAEs, from baseline through week 24 or week 25 through 52.
Discussion
Significantly greater proportions of patients receiving all tildrakizumab doses versus placebo achieved the primary endpoint of ACR20 at week 24. Among patients with prior anti-TNFα therapy, ACR20/50/70 treatment difference versus placebo was not apparent for all tildrakizumab dose arms, although there were relatively few such patients (n=17–19 per treatment arm). At week 24, PASI 75, 90 and 100 response rates were higher following treatment with all tildrakizumab doses versus placebo.
Definitive comparisons between the present results and other clinical studies cannot be made due to differences in trial design, study population and placebo response rates. However, proportions of ACR20/50/70 responders among patients receiving tildrakizumab 200 mg (Q4W and Q12W) were also numerically higher compared with previous trials of biologicals for PsA treatment.12–16 29–33 PASI 75/90/100 response rates were consistent with those reported in previous trials for tildrakizumab in psoriasis.22
MDA responses assess efficacy across the spectrum of PsA manifestations and are strongly associated with significant improvements in health-related QoL and productivity, making MDA an increasingly important treatment target in randomised PsA trials.34 In this study, significantly more patients receiving tildrakizumab versus placebo achieved MDA by week 24. PASDAS and DAPSA—additional established composite indices for measuring PsA disease activity27 35 36—were added as post hoc efficacy measures based on increasing recognition of their utility. Although statistical significance was not assessed for post hoc analyses, proportions of tildrakizumab-treated patients achieving DAPSA remission and PASDAS <3.2 at week 24 were higher relative to placebo-treated patients.
Tildrakizumab was generally well tolerated through week 52. Overall, safety findings were similar to the safety profile in phase III trials of tildrakizumab for treatment of plaque psoriasis (reSURFACE 1 and reSURFACE 2).22 There were no deaths or reports of systemic candidiasis, inflammatory bowel disease, MACE, or significantly increased liver enzymes through week 52.
Aberrant activation of the IL-23/IL-17 cytokine system is critical in the pathogenesis of PsA.37 IL-23 is thought to promote joint degeneration by inducing osteoclastogenesis and osteoclast-mediated activation of nuclear factor of activated T cells that regulate expression of genes facilitating pathological bone resorption (eg, matrix metallopeptidase 9).38 39 The efficacy and safety findings reported here may be attributed to selective antagonism of IL-23 by tildrakizumab. A plausible mechanism of action of tildrakizumab is inhibition of the IL-23-induced kinase signalling system resulting in reduced Th17 cell proliferation and downregulation of the Th17 cell-secreted inflammatory cytokines such as IL-17 and IL-22.10 37 39–41 By selectively targeting IL-23p19, tildrakizumab blocks IL-23-mediated signalling10 11 40 without targeting the p40 subunit common to both IL-23 and IL-12 (eg, ustekinumab). Tildrakizumab may thus circumvent potential adverse effects on cell immunity by sparing IL-12 function.22 42 43
Study limitations included high placebo response rates, confounding interpretation of results. Due to small numbers of patients in each subgroup, post hoc analyses detected no meaningful relationship between ACR20 placebo response at week 24 and patient baseline characteristics, background medication use, or country; however, placebo response rates were numerically lower in the USA and Spain relative to other countries. Per recent analyses, placebo response rates have increased over time in clinical trials across several disease states including rheumatoid arthritis, with no definitive explanation44–46; speculated causes in the rheumatoid arthritis study included expectation bias; therapeutic improvements resulting in a limited pool of eligible patients, possibly leading to recruitment during transient disease flares; and greater recruitment in resource-poor countries.44 Although placebo response was higher than expected, treatment difference and, therefore, statistical power were preserved. Relatively few patients with dactylitis or enthesitis were included, and baseline dactylitis in particular was not balanced among treatment arms, so the study was not powered to detect statistically significant differences in related endpoints. This is planned for attention in the phase III programme. The study was also not powered to differentiate tildrakizumab 100 and 200 mg doses. Mixed dose effects were observed. Patients treated with tildrakizumab 20 mg Q12W achieved greater improvement relative to placebo-treated patients for some efficacy measures, although response rates and improvement were smaller compared with patients receiving higher doses. Among patients receiving tildrakizumab 200 mg, Q4W dosing was not consistently superior to Q12W dosing. Patients receiving tildrakizumab 100 mg had numerically lower rates of ACR responses but numerically greater improvement in some component measures relative to those treated with tildrakizumab 200 mg. These findings were generally consistent with the small numbers of patients and the expected tildrakizumab dose–response relationship. Optimal dosing of tildrakizumab in patients with PsA is planned for investigation in the phase III programme.
Conclusion
These findings demonstrate that treatment with tildrakizumab 200 or 100 mg was more effective than placebo for rates of ACR20/50, DAS28-CRP, MDA and PASI 75/90/100 responses as well as improvement in physical function; effects were smaller for tildrakizumab 20 mg relative to higher doses. Tildrakizumab was well tolerated through 52 weeks of treatment. These results support tildrakizumab phase III clinical development in PsA.
Data availability statement
Data are available upon reasonable request. Data and other documents will be made available after publication, with no end date, to anyone who submits a reasonable request to the study sponsor.
Ethics statements
Ethics approval
The study protocol (online supplemental file) was approved by independent ethics committees or institutional review boards at each site. The trial was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Good Clinical Practice guidelines.
Acknowledgments
We thank the patients for their participation and Dr Siu-Long Yao of Sun Pharmaceutical Industries for study design and oversight. Some data were previously presented at the EULAR 2020 Annual European Congress of Rheumatology E-Congress beginning 3 June 2020 and published in the following congress abstracts: Mease PJ et al. Ann Rheum Dis 2020;79(Suppl 1):145; Gottlieb AB et al. Ann Rheum Dis 2020;79(suppl 1):1157; Nash P et al. Ann Rheum Dis 2020;79(Suppl 1):1167. Editorial support was provided by Puneet Dang, PhD; Claire Daniele, PhD, CMPP; and Judy Phillips, DVM, PhD, of AlphaBioCom, LLC, and funded by Sun Pharmaceutical Industries.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Lay summary
Disclaimer : This is a summary of a scientific article written by a medical professional (“the Original Article”). The Summary is written to assist non medically trained readers to understand general points of the Original Article. It is supplied “as is” without any warranty. You should note that the Original Article (and Summary) may not be fully relevant nor accurate as medical science is constantly changing and errors can occur. It is therefore very important that readers not rely on the content in the Summary and consult their medical professionals for all aspects of their health care and only rely on the Summary if directed to do so by their medical professional. Please view our full Website Terms and Conditions.
Copyright © 2021 BMJ Publishing Group Ltd & European League Against Rheumatism. Medical professionals may print copies for their and their patients and students non commercial use. Other individuals may print a single copy for their personal, non commercial use. For other uses please contact our Rights and Licensing Team.
Footnotes
Handling editor Josef S Smolen
Contributors All authors contributed to data interpretation and manuscript development, critically reviewed each draft for intellectual content and approved the final version for submission.
Funding The study was funded by Sun Pharma Global FZE. Analyses were funded by Sun Pharmaceutical Industries, Princeton, NJ, USA.
Competing interests PJM has received research grants from AbbVie; Amgen; Bristol Myers Squibb; Celgene; Janssen; Eli Lilly; Novartis; Pfizer; Sun Pharmaceutical Industries, Inc.; and UCB; consulting fees from AbbVie; Amgen; Bristol Myers Squibb; Boehringer Ingelheim; Galapagos; Gilead; GlaxoSmithKline; Janssen; Eli Lilly; Merck; Novartis; Pfizer; Sun Pharmaceutical Industries, Inc.; and UCB; and speaker fees from AbbVie, Amgen, Bristol Myers Squibb, Genentech, Janssen, Eli Lilly, Novartis, Pfizer and UCB. SC is a partner/physician at Arizona Arthritis and Rheumatology Associates. FJGF has received research grants, consulting fees and/or speaker fees from AbbVie, Eli Lilly, Gedeon Richter, MedImmune, Nichi-Iko, Pfizer, Sanofi-Aventis, Takeda and UCB. MEL has received research grants, consulting fees and/or speaker fees from AbbVie; Amgen; Eli Lilly; Genentech; Nichi-Iko; Novartis; Pfizer; R-Pharm; and Sun Pharmaceutical Industries, Inc. PR has received research grants from Janssen and Novartis, consulting fees from AbbVie, Amgen, Bristol Myers Squibb, Celgene, Janssen, Eli Lilly, Novartis, Pfizer, Roche and UCB; and speaker fees from AbbVie, Janssen, Eli Lilly, Novartis, Pfizer and UCB. SPR has received grants/research support from AbbVie; Janssen; Novartis; Pfizer; and Sun Pharmaceutical Industries, Inc.; and consulting fees from Amgen, Eli Lilly, Janssen, Novartis and Pfizer. RCC receives consultation fees from Sun Pharmaceutical Industries, Inc. AMM is a former employee of Sun Pharmaceutical Industries, Inc.; and has individual shares in Johnson and Johnson, and as part of retirement account/mutual funds. SJR is an employee of Sun Pharmaceutical Industries, Inc. ABG has received honoraria as an advisory board member and consultant for Avotres Therapeutics; Beiersdorf; Boehringer Ingelheim; Bristol-Myers Squibb Co.; Janssen; LEO Pharma; Eli Lilly; Novartis; Sun Pharmaceutical Industries, Inc.; UCB; and Xbiotech (stock options); and has received research/educational grants from Boehringer Ingelheim; Incyte; Janssen; Novartis; Sun Pharmaceutical Industries, Inc.; UCB; and Xbiotech (all paid to Mount Sinai School of Medicine).
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.