Article Text

Abstract
Objectives Recent advances in systemic sclerosis (SSc) show that it involves a T-helper type-2-oriented immune response with interleukin (IL)-4 and IL-13. Romilkimab is an engineered, humanised, bispecific immunoglobulin-G4 antibody that binds and neutralises IL-4/IL-13 making it ideal for exploration in fibrosis.
Methods Patients aged ≥18 years diagnosed with diffuse cutaneous SSc (dcSSc), and with or without immunosuppressive background therapy, were randomised (1:1) to subcutaneous romilkimab 200 mg or placebo one time per week for 24 weeks in this double-blind, proof-of-concept, phase II study. The primary endpoint was change in modified Rodnan skin score (mRSS) from baseline to week 24.
Results Ninety-seven patients were randomised to romilkimab (n=48) or placebo (n=49) for 24 weeks. Least-squares mean (SE) change in mRSS was –4.76 (0.86) for romilkimab versus –2.45 (0.85) for placebo yielding a mean (SE) (90% CI) difference of –2.31 (1.21) (–4.32 to –0.31; p=0.0291, one-sided). Treatment-emergent AEs were balanced between placebo (n=41; 84%) and romilkimab (n=40; 80%). Most were mild-to-moderate and discontinuations were low (three overall). There were two deaths (one scleroderma renal crisis (romilkimab) and one cardiomyopathy (placebo)), neither were considered treatment related. Two patients in the placebo group had a cardiovascular treatment-emergent SAE (one cardiac failure, one cardiomyopathy), but there were no cardiac safety signals with romilkimab.
Conclusion This study demonstrated significant effects on skin changes with romilkimab in early dcSSc that require confirmation with a longer and more comprehensive phase III study to determine clinical relevance.
Trial registration number NCT02921971.
- scleroderma
- systemic
- therapeutics
- chemokines
- autoimmune diseases
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Systemic sclerosis (SSc) is an orphan disease with no current treatments that can prevent disease progression.
There is strong evidence from clinical studies and animal models that interleukin (IL)-4 and IL-13 are involved in the pathology of SSc, but no therapies directed against these cytokines have been developed for the treatment of this disease.
What does this study add?
Romilkimab is a novel humanised bispecific immunoglobulin-G4 antibody that binds and neutralises both IL-4 and IL-13.
Results from this phase II proof-of-concept study showed that romilkimab achieved the primary endpoint (ie, a statistically significant reduction in modified Rodnan skin score at week 24) compared with placebo.
Romilkimab was well tolerated compared with placebo with no cardiac safety signals.
How might this impact on clinical practice or future developments?
This is the first study to highlight the potential of romilkimab in patients with diffuse cutaneous SSc (dcSSc), and its use warrants further investigation.
The study also provides further validation of the role that IL-4 and IL-13 may play in dcSSc.
Introduction
Systemic sclerosis (SSc) is characterised by immune dysregulation and microvascular obliteration in skin and internal organs.1 2 Overall prognosis remains poor, and diffuse cutaneous SSc (dcSSc) has a 10-year mortality rate of ~20%.3 4 Treatment choice is still debated. Nintedanib can reduce decline in forced vital capacity (FVC) in patients with SSc and lung fibrosis, and high-intensity regimens with autologous stem cell transplantation can favourably modify disease course in some patients.5–7
However, recent advances have shown that SSc involves a T-helper type-2 (TH2)-oriented immune response with key roles for interleukin (IL)-4 and IL-13.1 8 9 This profibrotic activity of TH2 cytokines is associated with increased periostin, a matricellular protein important in fibrogenesis.10 11 Furthermore, administration of anti-IL-4 and anti-IL-13 antibodies prevents the development of dermal fibrosis in animal models.2 8 This pathway, therefore, represents an exciting new avenue for fibrosis treatment.
Romilkimab (SAR156597) is an engineered humanised IgG4 antibody that binds and neutralises IL-4 and IL-13.12 13 Romilkimab reduced thymus and activation-regulated chemokine (TARC or chemokine (C-C motif) ligand 17) in patients with idiopathic pulmonary fibrosis (IPF); TARC is directly induced by IL-4 and IL-13 receptor activation, and a key serum marker used to assess target engagement.12 14 15 Romilkimab was well tolerated in healthy subjects who received single subcutaneous doses ranging from 10 to 300 mg. The safety profile was generally comparable between placebo, romilkimab 200 mg one time per week and once every 2 weeks in patients with IPF although serious adverse events (SAEs) were more common in the one time per week group (see online supplemental table S1 for safety outcomes).12 Romilkimab is a promising option given the pathology of SSc described.
Supplemental material
This 24-week, proof-of-concept study evaluated the efficacy and safety of romilkimab versus placebo in early dcSSc.
Methods
Design
This was a multinational, randomised, double-blind, placebo-controlled, proof-of-concept study that investigated romilkimab in patients with dcSSc. The study was conducted in 51 centres (39 randomised at least one patient) in 13 countries (Argentina, Belgium, Estonia, France, Germany, Italy, Mexico, Poland, Romania, Russian Federation, Ukraine, UK, USA). First patient was randomised on 21 December 2016 and last patient completed on 1 April 2019.
Patients were randomised (1:1) to receive romilkimab 200 mg one time per week subcutaneously or placebo one time per week subcutaneously. The study consisted of 4 weeks of screening, 24 weeks of treatment and 11 weeks of follow-up with no treatment (see online supplemental figure S1 for schedule). There was one protocol amendment (online supplemental table S3) driven by the imbalance of serious IPF and cardiac events between groups in the romilkimab study in IPF (online supplemental table S1).12 This amendment was endorsed by the independent Data Monitoring Committee (DMC). All patients provided written informed consent prior to participation.
Supplemental material
Participants
Patients were eligible if they were aged ≥18 years and were classified as having SSc,16 with the diffuse subtype as defined by LeRoy et al 17 criteria. Patients could enrol on stable background immunosuppressive therapies defined as: cyclophosphamide ≤1 mg/kg oral/day or ≤750 mg intravenously/month; azathioprine ≤100 mg/day; methotrexate ≤15 mg/week, and mycophenolate mofetil ≤2 g/day with or without low-dose corticosteroids (≤10 mg/day of oral prednisone or equivalent). See online supplemental file for exclusion criteria.
Randomisation, masking and treatment
Eligible patients were randomised to treatment via a centralised randomisation system using interactive response technology. Patients, investigators and study centre personnel (except those who reconstituted romilkimab and prepared syringes for injection) were blinded to treatment (online supplemental file).
Efficacy endpoints
Primary efficacy endpoint was change from baseline to week 24 in modified Rodnan skin score (mRSS; a validated measure of skin thickening18).
Secondary efficacy endpoints included change from baseline to week 24 in the Health Assessment Questionnaire-Disability Index (HAQ-DI), a measure of physical/functional disability, assessed using the Scleroderma Health Assessment Questionnaire (SHAQ)19–21 and observed FVC/observed diffusing lung capacity for carbon monoxide (DLCO; corrected for haemoglobin).
Exploratory efficacy endpoints included change from baseline to week 24 in the Visual Analogue Scale (VAS) for overall disease severity, pain severity, gastrointestinal function, breathing function, vascular function and digital ulcer impact on activity (assessed via SHAQ); University of California Los Angeles Scleroderma Clinical Trial Consortium Gastrointestinal Tract 2.0 total score22; tender joint count 28; digital ulcer count; and the European Quality of Life-5 Dimension-5 Level (EQ-5D-5L) index.23 Composite response index in diffuse cutaneous systemic sclerosis (CRISS) was measured at week 24 as defined in Khanna et al.24 Other exploratory endpoints included patients (%) with improvement in mRSS of at least 20%, 40% and 60% from baseline to week 24, mean change in mRSS in predefined subgroups (ie, more severe skin involvement at baseline (mRSS ≥15), disease duration <20 and ≥20 months, use of background medication (methotrexate, mycophenolate mofetil, azathioprine or cyclophosphamide), and medical history of SSc-interstitial lung disease (ILD) from baseline to week 24) and biomarkers of disease activity and the IL-4 and IL-13 pathway from baseline to week 24.
Post hoc analyses were undertaken to determine time to progression (ie, first event defined as death, relative change of at least 10% in % predicted FVC or at least 15% in % predicted DLCO (corrected for haemoglobin), increase of at least 20% or +5 in mRSS, or other defined Step 1 CRISS events not covered above), and patients with minimally important difference (MID) on mRSS calculated using the patients (%) that achieved an improvement in mRSS of 3.2 points at week 24. This MID estimate was based on a published study in SSc that used this threshold to illustrate clinically improved patients (effect size 0.40) at week 24.25
Safety endpoints
An independent DMC was responsible for reviewing safety data. Safety assessments consisted of recording adverse events (AEs), clinical laboratory evaluations, physical examinations, vital signs and ECGs. AEs were coded using Medical Dictionary for Regulatory Activities V.22.0. Potential for immunogenicity measured via antidrug antibody (ADA) response was also assessed.
Pharmacokinetics, immunogenicity and biomarker endpoints
Protein biomarkers associated with disease activity (cartilage oligomeric matrix protein, chemokine C-C motif ligand 2) and the IL-4/IL-13 pathway (TARC, periostin and eotaxin-3) were measured. Plasma samples tested for romilkimab were analysed by Bertin Pharma (Saclay, France). ADA testing was performed using a validated bridging qualitative ELISA with electrochemiluminescence detection (DSAR OC, Montpellier, France).
Statistical analyses
It was estimated that 94 patients (47 in each treatment group) would yield 80% power to detect a difference between romilkimab and placebo of 3.6 in the mean change from baseline in mRSS at 24 weeks, assuming an SD of 7 and using a one-sided alpha of 5% (type I error) to maximise the power needed to detect a positive signal with the limited sample size. This estimate was based on published studies.25 26
Efficacy endpoints were evaluated in the intent-to-treat population, defined as all randomised patients. Safety was reported in all randomised patients who received at least one dose or part dose of study treatment (safety population). Anti-romilkimab antibody analysis was performed in the safety population with at least one baseline and post-dose antibody sample. Efficacy endpoints were analysed using a mixed-effect model with repeated measures (MMRM) approach. Missing data were accounted for by the MMRM, which relied on the missing-at-random assumption. The model included interactions for fixed treatment effect, randomisation strata, timepoint and fixed baseline mRSS. The model provided baseline-adjusted least square (LS) means, SE and 95% CI. CRISS outcomes were compared between groups using van Elteren’s test stratified by randomisation strata.
Post hoc time to progression analysis was performed using a Cox model with mRSS at baseline, randomisation strata and treatment groups as explanatory variables. Post hoc analyses used two-sided tests. Romilkimab concentrations at selected timepoints after the last dose were reported using descriptive statistics. Pharmacokinetic parameters were estimated using the population pharmacokinetic approach at the lowest drug concentration before the next dose was administered (Ctrough). Analyses for safety and immunogenicity endpoints were descriptive.
Statistical analyses were performed using SAS (SAS Institute).
Patient involvement
Patients provided written informed consent at screening and attended scheduled clinic visits. Patients provided feedback regarding their condition throughout the study via the patient-reported outcome questionnaires.
Results
Participants
A total of 143 patients were screened, and 97 were randomised to treatment with romilkimab (n=48) or placebo (n=49). The main reason for screening failure in 46 patients was abnormal QuantiFERON-TB Gold results.
Eighty-seven (90%) randomised patients completed the 24-week treatment period and 10 (10%) patients permanently discontinued study treatment due to lack of efficacy (four, 4%), AEs (three, 3%) or other reasons (three, 3%) (online supplemental figure S2). Baseline demographic and clinical characteristics were generally comparable between treatment groups (table 1). Critical or major protocol deviations were reported in 9 (19%) patients from the romilkimab group and 19 (39%) patients from the placebo group. The higher rate of deviations in the placebo group was mainly related to randomisation procedures as shown in online supplemental table S4.
Supplemental material
Patient demographic and clinical characteristics at baseline
Primary efficacy endpoint
Romilkimab resulted in a statistically significant decrease in mRSS from baseline to week 24 versus placebo; the LS mean (SE) change was –4.76 (0.86) versus –2.45 (0.85), yielding a mean (SE) difference (90% CI) of –2.31 (1.21) (–4.32 to –0.31; p=0.0291, one-sided) (figure 1A). The observed mean (SD) mRSS at week 24 was 15.43 (7.12) for romilkimab (n=47) and 18.25 (8.62) for placebo (n=48).
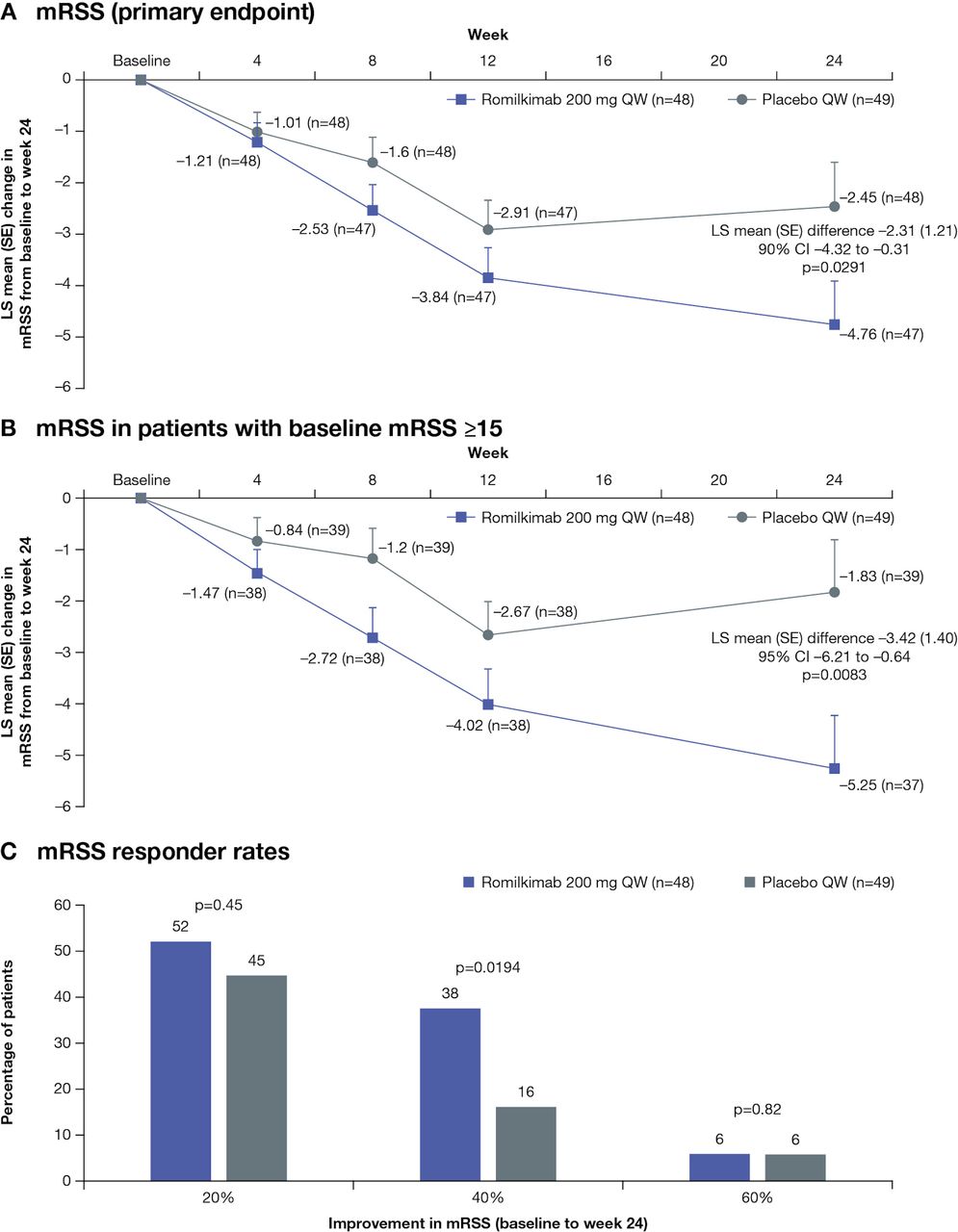
Mean change from baseline to week 24 in (A) mRSS (primary endpoint), (B) mRSS in more symptomatic patients at baseline (mRSS ≥15) and (C) mRSS responder rates (20%, 40% and 60% improvements in mRSS) in the ITT population treated with romilkimab versus placebo. ITT, intent-to-treat; LS, least squares; mRSS, modified Rodnan skin score; QW, one time per week.
Secondary efficacy endpoints
The LS mean (SE) change in FVC was –10 (40) mL for romilkimab versus –80 (40) mL for placebo at week 24 resulting in a non-significant mean (SE) difference (95% CI) of 70 (60) mL (–40 to 190; p=0.10) favouring romilkimab (figure 2A). The LS mean (SE) change for DLCO (haemoglobin corrected) was –0.12 (0.10) with romilkimab versus –0.27 (0.10) with placebo at week 24 yielding a non-significant difference of 0.15 (0.14) (–0.12 to 0.42; p=0.14) (figure 2B). The mean HAQ-DI score decreased slightly (ie, improved) in both groups (figure 2C). The LS mean (SE) change was –0.09 (0.08) for romilkimab versus –0.12 (0.08) for placebo at week 24; the LS mean (SE) difference (95% CI) was 0.03 (0.11) (–0.19 to 0.24; p=0.40).

Mean change from baseline to week 24 in (A) FVC (mL), (B) DLCO and (C) HAQ-DI in the ITT population treated with romilkimab versus placebo. DLCO, diffusing lung capacity for carbon monoxide; FVC, forced vital capacity; HAQ-DI, Health Assessment Questionnaire-Disability Index; ITT, intent-to-treat; LS, least squares; QW, one time per week.
Exploratory efficacy endpoints
Romilkimab resulted in a statistically significant improvement in the EQ-5D-5L index compared with placebo; the LS mean (SE) change from baseline to week 24 was 0.07 (0.03) for romilkimab versus 0.00 (0.03) for placebo resulting in a mean (SE) difference (95% CI) of 0.07 (0.04) (–0.01 to 0.15; p=0.0363) (table 2). Additional exploratory efficacy endpoints are summarised in online supplemental table S5.
Mean change from baseline to week 24 in exploratory patient-reported outcomes in the ITT population treated with romilkimab versus placebo
Prespecified subgroup analyses
The LS mean (SE) difference (95% CI) in mRSS was statistically significantly in favour of romilkimab versus placebo in patients with more severely affected skin (ie, baseline mRSS ≥15; –3.42 (1.40) (–6.21 to –0.64; p=0.0083)) (figure 1B). Responder rate analysis indicated that 20%, 40% and 60% improvements in mRSS from baseline to week 24 was higher for romilkimab than placebo; the between-group difference for 40% improvement in mRSS was statistically significant (p=0.0194) (figure 1C). The LS mean difference in mRSS was numerically in favour of romilkimab versus placebo at week 24, regardless of the baseline disease duration (<20 and ≥20 months), use of background therapy or medical history of SSc-ILD (online supplemental table S6).
Post hoc analyses
There was a trend of benefit for romilkimab in time to an event reflecting disease progression compared with placebo: 9 (19%) versus 15 (31%) (HR 0.47 (95% CI 0.20 to 1.11); p=0.09, two-sided), respectively (online supplemental figure S3 and table S7). Regarding skin thickness, we showed that a larger proportion of patients achieved an MID threshold of 3.2 units on mRSS change from baseline in the romilkimab than placebo group (54% vs 43% (90% CI 0.82 to 3.28)), respectively.
Supplemental material
Safety endpoints
Although the overall incidence of treatment-emergent AEs (TEAEs) was high (>80% in both groups, n=41 for placebo and n=40 for romilkimab), most were mild or moderate in intensity (n=19 (40%) mild, n=20 (42%) moderate and n=1 (2%) severe for romilkimab; and n=23 (47%) mild, n=14 (29%) moderate and n=4 (8%) severe for placebo), and the incidence of TEAEs leading to permanent treatment discontinuation was low (three overall) (table 3). There were two deaths (one scleroderma renal crisis on romilkimab and one cardiomyopathy on placebo), but neither were considered treatment related by the investigator. There were no clinically meaningful changes in laboratory parameters, vital signs or ECGs over the course of the study.
Summary of patients experiencing AEs over the 24-week study
Pharmacokinetics, immunogenicity and biomarker endpoints
Pharmacokinetic analysis showed that steady state for romilkimab was reached by week 4. Arithmetic mean (SD) Ctrough was 38.23 (17.96) μg/mL at week 4 and 47.45 (30.23) μg/mL at week 24. Immunogenicity testing showed that three patients in the romilkimab group and zero in the placebo group developed positive ADAs by week 24; all were considered low titre (30). None were associated with TEAEs.
Romilkimab was associated with a statistically significant reduction in TARC versus placebo, indicating a positive effect on this pathway; the LS mean difference (95% CI) at week 24 was –115.56 ng/L (–216.87 to –14.26; p=0.0258) (figure 3A). Periostin also showed a strong trend for greater decline with romilkimab versus placebo (figure 3B). Additional biomarkers were not significantly different between romilkimab and placebo (online supplemental table S8).

{kind=link}
{kind=link}
{kind=link}
Mean change from baseline to week 24 for (A) TARC and (B) periostin in patients treated with romilkimab versus placebo. LS, least squares; QW, one time per week; TARC, thymus and activation-regulated chemokine.
Discussion
This is the first study to evaluate the efficacy and safety of romilkimab in patients with early dcSSc. Importantly, this phase II proof-of-concept study showed that romilkimab was associated with a statistically significant reduction in skin fibrosis versus placebo. Predefined subgroup analysis indicated that the efficacy of romilkimab on mRSS extended to patients with the most severe disease at baseline (mRSS ≥15), and it was effective in patients at the earliest disease stages of less than 20 months.
Although MID estimates for mRSS are not available at week 24 in other recently completed studies,27 28 a previous analysis from the D-penicillamine study suggested an estimate of 3.2 units at week 24.25 Applying this estimate to our study resulted in a larger proportion of patients achieving MID with romilkimab versus placebo although not reaching statistical significance. Altogether, these data are consistent with a meaningful benefit of romilkimab on skin involvement but should be interpreted with caution in a phase II study. Therefore, future studies should incorporate appropriate anchors to calculate the MID estimates at the 24-week period with background standard of care therapy.
Comparisons with other studies are favourable. FaSScinate was a phase II, 48-week study of tocilizumab in SSc.28 In this study, tocilizumab was associated with a numerically greater improvement in mRSS than placebo at 24 weeks; the LS mean change was ‒3.92 versus ‒1.22 (p=0.09).28 In our study, there was a statistically significant change in patients that would have been eligible for faSScinate (ie, those with higher baseline mRSS of ≥15). It is important to note, however, that background therapy was allowed in both groups in our study but was not allowed in faSScinate, and this may have contributed to the greater mRSS change in the placebo group. There was also indication of a possible additive effect of romilkimab in patients receiving background immunosuppressive therapy (56%; n=54/97) as shown by the change in mRSS at week 24 compared with patients without background medication (‒5.81 vs ‒3.64, respectively). Herein, there was an initial improvement of mRSS up to week 12 in both groups, followed by slight worsening of mRSS up to week 24 in the placebo group, which may reflect the initial impact of background therapy. However, the study was not designed to examine prior medication use in detail. Nevertheless, allowing background immunosuppressant in a study looking primarily at skin outcomes was novel. Our compelling results together with those of the SENSCIS lung study5 should establish a new standard for SSc studies by allowing background immunosuppressants. Another randomised study (ASSET) showed that abatacept was associated with a numerically greater improvement in adjusted mRSS (primary endpoint) than placebo at 12 months (mean (SE): ‒6.24 (1.14) vs ‒4.49 (1.14); p=0.28), but this study failed to reach statistical significance.29 It is not possible to determine if different mechanism of action between agents accounted for the variable results, but it must be noted that the primary analyses in our study were based on prespecified one-sided compared with two-sided alpha testing in faSScinate and ASSET. Despite several recent studies of potential therapies not meeting the mRSS endpoint, skin score assessment remains clinically relevant, alongside organ-specific or patient-reported outcomes in defining clinical benefit in SSc studies. Future studies may use combined composite endpoints to more robustly evaluate efficacy in SSc.
In the current study, romilkimab had a non-significant but favourable effect on lung outcomes, which warrants further evaluation. The loss of 80 mL for FVC between baseline and week 24 for patients treated with placebo supports previous findings that patients with early dcSSc may develop significant lung disease.30 Additionally, this highlights an overlap between the immunopathology of dcSSc and IPF, but there may be mechanistic differences between these two fibrotic conditions.
Safety results were generally similar to those previously reported for patients who received romilkimab for the treatment of IPF.12 However, no serious or severe cardiac or respiratory TEAEs were reported in this study for those treated with romilkimab. Oral herpes has been reported with other IL-4/IL-13 blocking agents. Herein, oral herpes was more common in romilkimab-treated patients, but no herpes zoster or disseminated herpes infection was observed.
This study is limited by a relatively short treatment duration, which may not have permitted the detection of significant differences in other non-skin outcomes; however, this was consistent with previous phase II studies, and was chosen based on rapid response in skin outcomes, seen in other studies.28
In conclusion, this is the first study of romilkimab in patients with early dcSSc, and our findings provide strong justification for further studies. The impact on skin met the prespecified endpoint, which is the first time that this has been achieved in a phase II study in dcSSc.
Acknowledgments
Writing assistance was provided by Sam Phillips, PhD, Parexel, Uxbridge, UK, and was funded by Sanofi. DMC members included Christian Pagnoux, MD, MSc, MPH (Chairman); Ami Shah, MD, MHS; and Lucy Shneyer, MA. The authors would also like to thank the patients who participated in this study, and their families, as well as the investigators, study coordinators, study teams and nurses. Centre details are listed in online supplemental table S9.
References
Footnotes
YA and PW are joint first authors.
Handling editor Josef S Smolen
Collaborators Yannick Allanore, Hugo Ascencio, Shervin Assassi, Hilario Avila Armengol, Jerome Avouac, Andra Balanescu, Guillermo Gerardo Bartel, Doreen Belz, Lorenzo Beretta, Horacio Berman, Francesco Boin, Mihai Bojinca, Violeta Bojinca, Martin Brom, Justyna Cal-Kocikowska, Maria Gabriela Cardozo, Renato Carignola, Emmanuel Chatelus, Soumya Chatterjee, Dorota Cieslak, Maurizio Cutolo, Maria Dall’Era, Behrus Darvishan, Ellen De Langhe, Nicoletta Del Papa, Christopher Denton, Oxana Desinova, Villafañe Nicolas Javier Destefani, Katrin Elsharkawi-Welt, Jorge A Esquivel-Valerio, Gulshat Fatkhullina, Dmytro Fedkov, Eugen Feist, Renaud Felten, Rosanna Ferreira Lopez, Alvarado Diana Elsa Flores, Rene A Flores-Franco, Ignacio Garcia-De La Torre, Maria Victoria Garcia, Ignacio Garcia Valladares, Olena Garmisch, Ana Gheorghiu, Andrey Gnilorybov, Bello Yelitza Concepción González, Maria Groseanu, Walter Hermann, Pawel Hrycaj, Oleg Iaremenko, Janusz Jaworski, Christian Jorgensen, Rimma Kamalova, Alina Kamynina, Claudia Kedor, Natalie Kelp, Elsa Khazeeva, Piotr Klepacki, Aleksandra Kolczewska, Ramona König, Volodymyr Kovalenko, Thomas Krieg, Marina Letaeva, Pavlo Logoida, Natalia Loschits, Lukasz Lubinski, Wanda Maglione, Olga Malyshenko, Elena-Laura Margarint, Ciprian M Marinescu, Alejandro Martelli, Maureen Mayes, Diana Mazilu, Valeriia Melokhina, Karin Melsens, Christopher Meneses Lopez, Maria Mirza, Maria Misterska-Skora, Pia Moinzadeh, Erika Montabone, Ulf Müller-Ladner, Edgardo Munoz-Esteves, Ene Ojassalu, Daniela Opris, Cesar Tena Pacheco, Hugo Parra Ruiz, Lorena Perez Barbosa, Yves-Marie Pers, Ana Petcu, Liubov Petelytska, Christiane Pfeiffer, Yves Piette, Oksana Pirogova, Horatiu V Popoviciu, Kaitlin Quinn, Tatiana Raskina, Eve-Kai Raussi, Simona Rednic, Greta Reyes, Janett Riega, Roberto Jesus Gallegos Rivero, Mariano Rivero, Andrii Romanovskyi, Ewa Rzeszewska, Mirtha Sabelli, Mikhail Sandin, Maria Lida Santiago, Mikk Saretok, Ioana Saulescu, Lena Schriever, Agata Sebastian, Adriana Severino, Anca Sindrilaru, Cassandra Skinner, Vanessa Smith, Christelle Sordet, Tatiana Sotnikova, Jorge Alberto Spindler, Javier Walter Spindler, Marina Stanislav, Gerald Stapfer, Mayya Starovoytova, Virginia Steen, Simona Szasz, Guillermo Tate, Patricio Tate, Piia Tuvik, Adelfia Quezada Urenda, Lyalya Valishina, Amber Vanhaecke, Brenda Vázquez, Ildiko Vicsi, Barbara Vigone, Oana Vutcanu, René Westhovens, Victoria Zimina, Anna Zubrzycka-Sienkiewicz. Primary investigators are highlighted in bold.
Contributors YA and CPD contributed to the study concept and design, and data collection, analysis and interpretation. PW contributed to the study concept and design, data analysis and interpretation. DK, CE, FM and AL contributed to data analysis and interpretation. CS contributed to the study concept and design and data interpretation. RB designed the study analyses and contributed to data interpretation. All authors contributed to manuscript preparation and approved the final version.
Funding The study was funded by Sanofi R&D, France.
Competing interests YA reports personal fees from Actelion, Bayer, BMS and Curzion, and grants and personal fees from Inventiva, Roche and Sanofi. CPD reports personal fees from Actelion, Bayer, Boehringer Ingelheim, Corbus, Roche, Sanofi, and grants and personal fees from CSL Behring, GlaxoSmithKline and Inventiva. DK reports personal fees from Acceleron, Actelion, Blade Therapeutics, Eicos Sciences, Inc, Galapagos, GlaxoSmithKline, Mitsubishi Tanabe, Sanofi-Aventis/Genzyme and UCB Pharma, and grants and personal fees from Bayer, Bristol Myers Squibb and Genentech/Roche; he is funded by the NIH/NIAMS K24 AR063120. PW, CS, CE, FM, RB and AL are employees of Sanofi. CS is a shareholder in Sanofi.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Obtained.
Ethics approval The study was conducted in accordance with Declaration of Helsinki and International Conference of Harmonisation guidelines for Good Clinical Practice. Each participating site’s institutional review board or ethics committee approved the protocol and amendment(s) (online supplemental table S2).
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request. Qualified researchers may request access to patient level data and related study documents, including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan and dataset specifications. Patient level data will be anonymised, and study documents will be redacted to protect the privacy of study participants. Further details on Sanofi’s data sharing criteria, eligible studies and process for requesting access can be found at: https://www.clinicalstudydatarequest.com.