Article Text

Abstract
Objectives We explored the effects of B-cell directed therapy in subjects at risk of developing autoantibodypositive rheumatoid arthritis (RA), who never experienced inflammatory arthritis before, and explored biomarkers predictive of arthritis development.
Methods Individuals positive for both anti-citrullinated peptide antibodies and rheumatoid factor but without arthritis were included in a randomised, double-blind, placebo-controlled study to receive a single infusion of 1000 mg rituximab or placebo.
Results Eighty-one individuals received treatment and were followed up for a mean of 29.0 (0–54) months, during which 30/81 (37%) individuals developed arthritis. The observed risk of developing arthritis in the placebo-treated group was 40%, which was decreased by 55% (HR 0.45, 95% CI 0.154 to 1.322) in the rituximab-treated group at 12 months. Rituximab treatment caused a delay in arthritis development of 12 months compared with placebo treatment at the point when 25% of the subjects had developed arthritis (p<0.0001). Erythrocyte sedimentation rate and the presence of anti-citrullinated α-enolase peptide 1 at baseline were significant predictors of arthritis development.
Conclusions A single infusion of 1000 mg rituximab significantly delays the development of arthritis in subjects at risk of developing RA, providing evidence for the pathogenetic role of B cells, prearthritis stage of autoantibody positive RA.
- prevention
- cure
- rheumatoid arthritis
- rituximab
- pre-rheumatoid arthritis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known about this subject?
From earlier research reports we learnt that rheumatoid factor (RF) and Anti-Citrullinated Peptide Antibodies (ACPAs) can be found in the peripheral blood of individuals >10 years before the development of autoantibody positive rheumatoid arthritis.
Research leading to the recognition of this phase of systemic autoimmunity has not only supported the view that the pathogenetic process might not be initiated in the joint but created an opportunity to potentially delay the clinical onset of disease by a targeted intervention in this early phase.
B-cells play a pivotal role in this process as apart from being predecessors of cells that produce immunoglobulins including RF and ACPAs, B-cells are efficient antigen presenting cells, may activate T cells in the context of co-stimulatory signals, and produce a variety of cytokines.
Indeed, B-cell targeted therapy is effective in early as well as in late established RA.
Added value of this study
With a targeted intervention aimed at eliminating a cell key to the underlying pathogenetic process, the B cell, and influencing their function and products, the results of this study support the concept of a preventive window of opportunity.
In an exploratory randomised, double-blind, placebo controlled clinical trial, we show that a single infusion of 1000 mg of rituximab delays the onset of clinical signs and symptoms of arthritis in subjects who are at a high risk of developing seropositive RA.
How might this impact on clinical practice or future developments?
According to the current treatment paradigm, treatment of RA is initiated after the clinical onset of the disease.
With this approach only a small minority of patients achieve disease remission, which is the treatment goal, and many patients need chronic treatment with biopharmaceuticals or targeted small molecules.
The results of this study support the view that it may be easier to control the disease process by targeted intervention before signs and symptoms of arthritis have developed, which suggests the existence of a ‘preventive window of opportunity’.
Experimental interventions during the earliest stages of immune-mediated inflammatory diseases may provide important insights into their pathogenesis. Autoantibody positive rheumatoid arthritis (RA) is a common and prototypic autoimmune disease. This condition can be preceded by a phase of systemic autoimmunity during which circulating autoantibodies, increased acute phase reactants, proinflammatory cytokines and chemokines are found, even years before the development of clinically evident arthritis.1–4 Elevated levels of autoantibodies such as IgM rheumatoid factor (IgM-RF), anti-citrullinated peptide antibodies (ACPA) and other RA-specific antibodies against post-translationally modified proteins can be detected in blood samples of individuals later diagnosed with seropositive RA with a median of 5 years before arthritis becomes evident.3 During this stage, clonal changes in the peripheral blood B-cell receptor (BCR) repertoire can be detected5 but the synovial tissue is usually completely normal.6 7 The risk of developing arthritis within 2 years in individuals positive for both ACPA and IgM-RF is ~40%.8 This risk appears to be higher in individuals with musculoskeletal symptoms,9 smokers,10 in people who are obese10 and in those with decreased vagus nerve tone.11 The contribution of the HLA-DRB1 alleles encoding the shared epitope to RA development is mainly mediated via the presence of ACPA and does not appear to be a strong predictor of RA development within the ACPA positive pre-RA population.12 The existence of this preclinical phase offers the opportunity to intervene, and prevent or delay the disease from developing into clinically manifest arthritis.13 14
The presence of circulating autoantibodies and changes in BCR repertoire years before the clinical onset of the disease, the specificity of ACPA for the diagnosis of RA and the presence of B cells and plasma cells at the site of inflammation in early established disease15 highlight the importance of B cells in the pathogenesis of RA. Indeed, treatment of patients with RA with depleting antibodies directed at B cells is effective in late as well as earlier stages of established RA.16–19 However, there is no experimental evidence for B cells as therapeutic or secondary preventive target during the prearthritis stage of this autoantibody positive immune-mediate inflammatory disease. The purpose of this phase IIb, randomised, double-blind, placebo-controlled study was to test whether B-cell depletion could alter the development of the disease in individuals at high risk of developing RA. We also aimed to identify biomarkers predictive of arthritis development.
Methods
Participants and study design
One hundred and nine subjects with arthralgia8 20 21 without any evidence of clinical arthritis (of 66 joints examined) were recruited via rheumatology outpatient clinics of seven participating centres across the Netherlands between January 2010 and December 2013, of which 82 were eligible to be randomised and included in this multicentre, randomised, double-blind, placebo-controlled clinical study (The PRAIRI study: Prevention of clinically manifest rheumatoid arthritis by B-cell directed therapy in the earliest phase of the disease, NTR1969) (figure 1). The original aim of recruiting 90 eligible subjects was amended due to a slow recruitment rate encountered during the third year of the study, still keeping within the original power calculations. Ways of recruiting potential subjects included subjects referred via their general practitioner, engagement of first-degree relatives of known patients with RA via the outpatient clinic (Academic Medical Centre (AMC), Amsterdam) and first-degree relatives recruited at public fairs across the Netherlands: the proband diagnosis of RA was determined by questioning of the first-degree relative by a trained physician who attended the fair. These potential subjects were invited to be screened at the outpatient clinics of the participating centres. To be included into the study, the subjects needed to be between 18 and 80 years old, IgM-RF as well as ACPA (a-CCP2; Immunoscan CCPlus (Euro Diagnostica No RA-96plus) ELISA tests) positive and had to never experienced an inflammatory arthritis nor been treated with a disease-modifying antirheumatic drug (DMARD) in the past (phase c+d of the preclinical phase of RA).22 In addition, the subjects needed to have either C-reactive protein (CRP) levels >0.6 mg/L at screening (the lower limit of detection of the high-sensitivity (hs) CRP assay), or subclinical synovitis as determined by ultrasound or MRI using gadolinium performed in the context of routine clinical care. The cut-off level for serum CRP levels of 3 mg/L in the protocol published on the NTR website was amended into a minimum level of 0.6 mg/L, a change triggered by the advent of the hsCRP assay in routine clinical practice, as the target study population does not have overt inflammation during the preclinical stage of the disease. This amendment was approved by the Medical Ethics Committee of the AMC before enrolment of any subject into the study, who all provided informed consent. The subjects were randomised in a 1:1 ratio to receive either 1000 mg of rituximab (MabThera, Roche Nederland) or placebo (NaCl 0.9%) intravenously, after all receiving 100 mg methylprednisolone premedication according to the regular treatment schedule used in patients with RA to prevent potential infusion-related adverse events. Randomisation was stratified for age (<40 years, ≥40 years) as well as gender. One individual withdrew informed consent before receiving study treatment. The primary outcome was time to development of clinical arthritis in subjects in both treatment groups. Clinical arthritis was defined by a swollen and tender joint as observed by two independent, blinded investigators (one rheumatological research physician well trained in assessing joints in clinical trials and one faculty rheumatologist); consensus was reached after assessing the joint together in case of initial discrepancy (for details on the amended in and exclusion criteria compared with the NTR registration information and sample size calculation, visit scc, see online supplementary file). The study physicians, monitors and subjects remained blinded during the study, and all assessments were done by assessors blinded to the treatment allocation. The members of an independent data safety monitoring board and one independent physician overseeing laboratory results for safety reasons were unblinded to the treatment allocation.
Supplemental material

Trial profile. CRP, C-reactive protein; RF, rheumatoid factor.
Explorative analysis of the effects of study treatment on peripheral blood T and B-cell numbers, their subpopulations using fluorescence-activated cell sorting (FACS) analysis and the presence and levels of disease-specific antibodies were measured in subsets of participants depending on the availability of the samples for the different time points. We measured serum antibodies against various citrullinated peptides and arginine-containing peptides, including anti-alpha citrullinated enolase peptide-1 (CEP-1). The difference between citrullinated and arginine peptides was calculated and the cut-off level defining positivity for each ACPA specificity was determined on the basis of the earlier determined 98th percentile.7 Absolute levels (arbitrary units, AU) calculated from a calibration sample were used to follow individual and mean changes over time (details on the detection of other autoantibodies against citrullinated peptides can be found in the online supplementary file).
Statistical analysis
All subjects who received treatment were included in the primary and safety analysis. Kaplan-Meier survival analysis was used to determine the effect of rituximab treatment on the development of arthritis. Whether the rituximab treatment effect on the hazard to develop arthritis varied with follow-up time was evaluated using Cox proportional hazards regression by including the interaction between treatment and follow-up time as a continuous time-dependent variable in the model. The Cox model was also used to evaluate the effects of baseline patient characteristics and biomarkers on the hazard to develop arthritis. The change patterns over time during the study of time-dependent biomarkers were analysed using linear mixed-effects regression models with follow-up time, treatment and their interaction as fixed effects and with random intercept and slope(s) of follow-up time per patient as random effects. Joint models were used to evaluate the associations between the changing values over follow-up time of the time-dependent biomarkers and the arthritis hazards. Included in these joint models (a combination of the Cox and mixed-effects models) as a covariate were the predicted values of the time-dependent biomarkers of all individuals at risk for developing arthritis at all time points during follow-up. These joint models were evaluated for each biomarker separately. SPSS V.22 (SPSS), SAS V.9.3 and R V.3.3.2 were used to analyse the data.
Results
No significant differences in demographic and clinical features such as age, gender, inflammatory markers in the peripheral blood, tender and swollen joint counts, levels of IgM-RF or ACPA, smoking history, body mass index (BMI) and use of non-steroidal anti-inflammatory drugs (NSAID) were observed between the groups at baseline (table 1). None of the subjects had a swollen joint at baseline per protocol. All included subjects met the inclusion criteria based on laboratory parameters and none underwent imaging to ascertain inclusion status. However, in the context of routine care, a subset of 48 subjects had undergone imaging of their joints prior to the study of which 46 did not show synovitis in those joints.
Baseline characteristics of subjects assigned to the two treatment groups
Response to treatment
After treatment of 81 subjects (41 received rituximab and 40 placebo), follow-up of a median of 29 months (IQR 14–40; range 0–54 months; one subject developed arthritis 3 weeks after treatment) was available. The risk of development of arthritis over the total follow-up time in the placebo group was 40%. From the routine characteristics measured at baseline, including all baseline characteristics mentioned in table 1: gender, age, CRP, erythrocyte sedimentation rate (ESR), Patient Global Assessment of Disease Activity, tender joint count, IgM-RF presence as well as high and low positive levels, ACPA presence as well as high and low positive levels, BMI, smoking history (ever or never) and current NSAID use, only ESR was correlated with the development of arthritis (p=0.02). Otherwise, no statistically significant differences were found between the subjects who developed arthritis and those who did not. Power calculations were not performed on these baseline characteristics before study start.
Treatment with only one single infusion of rituximab reduced the baseline risk of arthritis development observed in the placebo group with 55% at 12 months (HR 0.45, 95% CI 0.15 to 1.32; p=0.15; see figure 2) and 53% at 18 months (HR 0.48, 95% CI 0.19 to 1.19) of follow-up. The treatment led to a delay of arthritis development of 12.0 months at the point where 25% of the subjects in both treatment groups developed arthritis (the 25th percentile or 75% free of arthritis of the cumulative arthritis-free survival;12 months placebo vs 24 months rituximab). A Cox proportional hazards model was used to analyse the data with treatment and treatment by follow-up time interaction, confirming the statistically significant although temporary preventive effect of rituximab treatment (p<0.0001). The observed effect on delaying arthritis development attenuated over time. Over the complete follow-up time arthritis development was seen in 30 of the 81 subjects: 16/40 (40%) in the placebo group after a median period of 11.5 months (IQR 2.5–15.0, range 1.0–40.0 months) and 14/41 (34%) in the rituximab group after a median period of 16.5 months (IQR 9.0–28.0, range 1.0–37.0 months). The risk of arthritis development over the total follow-up time after a single infusion was not statistically significant (p=0.448) between the two groups.
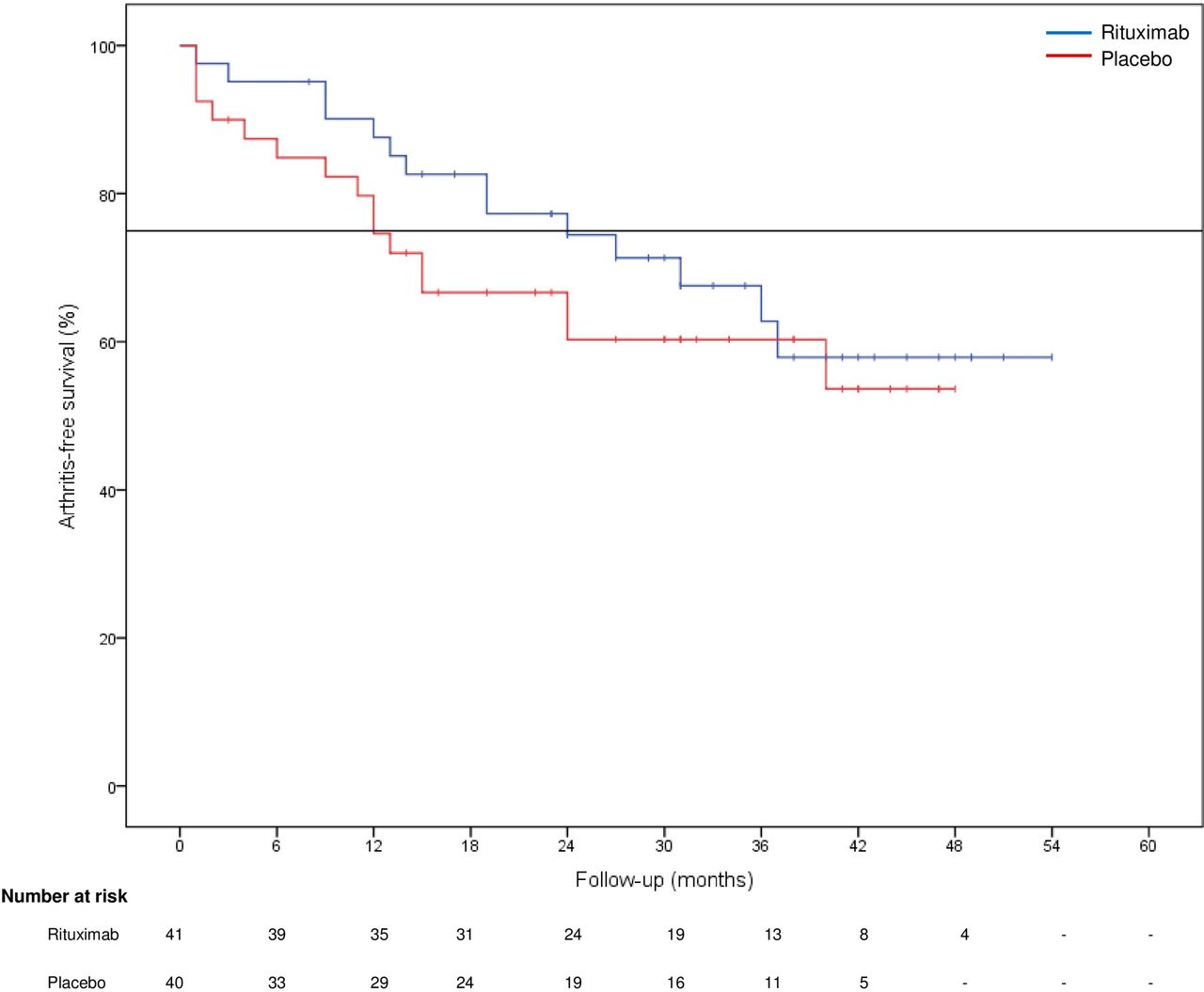
Kaplan-Meier survival plot for primary endpoint of arthritis development. Arthritis-free survival (%) depicted over time in months. At the 25th percentile, a difference of 12 months between the group receiving placebo (blue) versus rituximab (red) was observed (black horizontal line). The number of individuals at risk in each group at every follow-up time point is shown below the graph, follow-up was discontinued after development of arthritis.
At the moment of arthritis development, 13 subjects in the rituximab treatment group fulfilled the American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) 2010 classification criteria for RA,23 whereas one patient was classified as having unclassified arthritis.22 Of the 16 subjects in the placebo group who developed arthritis, 11 fulfilled the ACR/EULAR classification criteria for RA at the time of arthritis development, while five subjects were classified as having unclassified arthritis based on the low number of clinically inflamed joints and low levels of ESR and CRP at the time of arthritis development; three of these were classified as RA after further follow-up. Overall, four subjects without arthritis were lost to follow-up (two in each treatment group).
Exploratory analysis of biomarkers
The association of baseline and repeatedly measured clinical and serological biomarkers with the arthritis hazard was evaluated based on data of all 81 patients. Of these markers, the ESR (mm/hour; HR 1.03, 95% CI 1.01 to 1.06; p=0.016) and the presence of anti-citrullinated α-enolase peptide 1 (anti-CEP-1; HR 3.71, 95% CI 1.51 to 9.18; p=0.01) in the serum at baseline were positively correlated with the development of arthritis (Supplementary file 1).
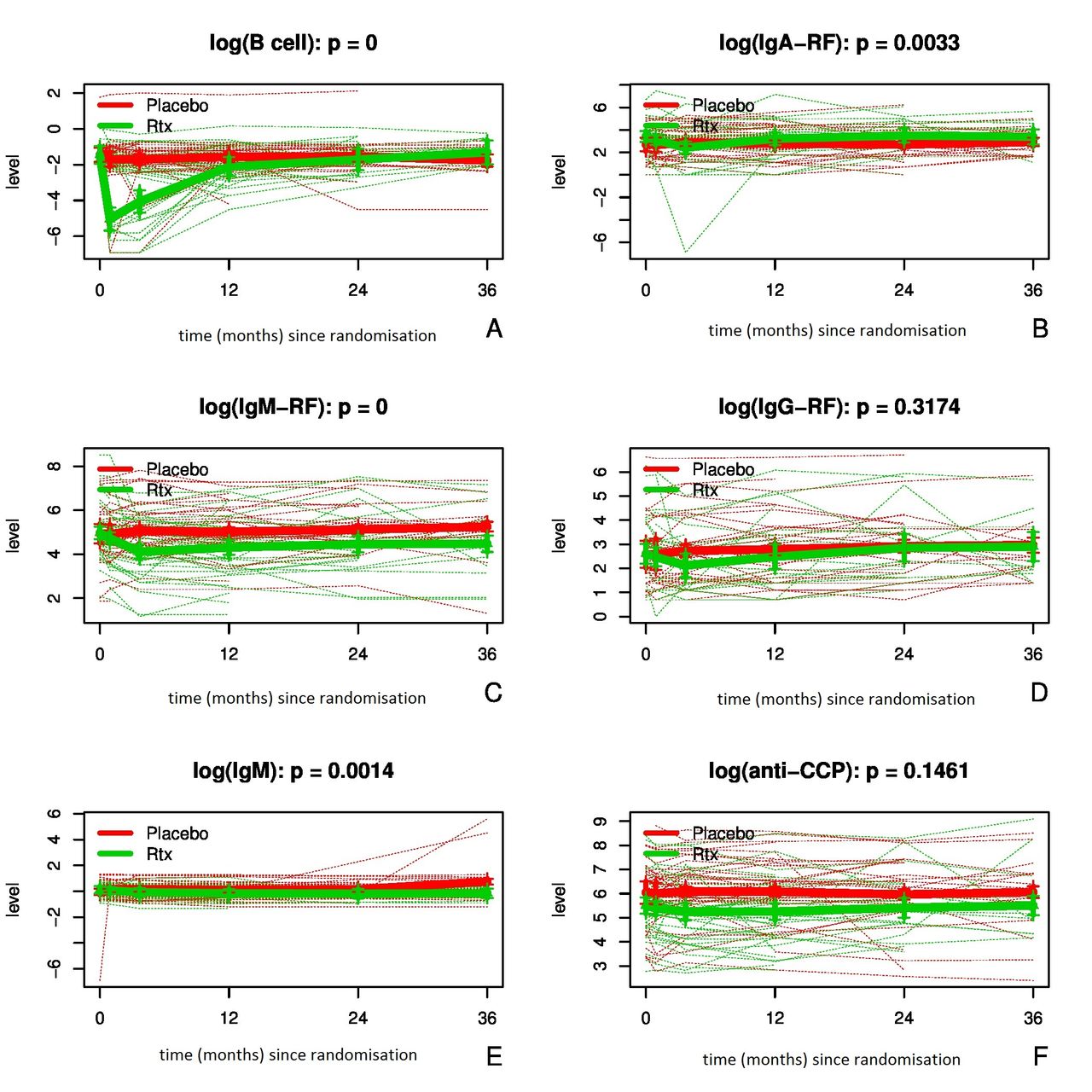
Changes in total B-cell numbers (×109/L), their subsets and disease relevant autoantibodies were available in a subgroup of 78 individuals (of whom 40 received rituximab and 38 placebo treatment) from baseline up to 3 years of follow-up. In addition, in a smaller subgroup (n=47; n=19 rituximab and n=28 placebo) serum levels of IgA-RF (kU/L), IgG-RF (kU/L), IgM-RF (kU/L), IgA (g/L), IgG (g/L), total IgM (g/L) as well as anti-citrullinated cyclic peptide (anti-CCP; kAU/L) could be measured at the same time points. A clear and highly significant decrease in the total number of B cells was observed within 4 weeks after treatment in the subjects receiving rituximab (p<0.0001), which was followed by a drop in serum levels of IgA-RF, IgM-RF and IgM, reaching the level of statistical significance (p=0.003, p<0.0001 and p=0.001, respectively) at all time points. Anti-CCP and IgG-RF serum levels dropped as well, but there was no statistical difference between the two treatment groups (p=0.146 and p=0.317, respectively; all values see figure 3A–F). No differences in the levels of total IgA and IgG between the two treatments were found.

{kind=link}
{kind=link}
{kind=link}
(A–F) Changes of B-cell numbers and B-cell related biomarkers. Total number of B cells (Log10 109/L; A), serum IgA rheumatoid factor (RF) (Log kU/L; B), IgM-RF (Log kU/L; C), IgG-RF (Log kU/L; D); IgM (Log g/L; E), and anti-citrullinated cyclic peptide (CCP) test levels (Log kAU/L; F) from baseline to follow-up time points (days) measured in the individuals treated with placebo (red) and rituximab (green). The thin lines represent changes in the individuals and the thick lines represent the mean numbers/levels for each group. Vertical lines represent the 95% CIs. Statistically significant differences (shown p values) between the two treatment groups were found for all values depicted here, except for the serum IgG-RF and anti-CCP levels.
The changes in serum levels of IgA-RF, IgM-RF and IgM in the group of individuals treated with rituximab were not associated with the development of arthritis.
A more detailed analysis of subsets of B-cell populations using FACS analysis was performed in 45 subjects (n=18 rituximab, n=27 placebo), based on availability of samples. The results of these analyses can be found in the online supplementary figures S1 and S2.
Safety of the treatment
Study treatment was generally well tolerated with only mild infusion-related symptoms and no serious infections leading to hospitalisation. Although the serious adverse event rate was significantly higher in the rituximab group (13/41 vs 3/40: p=0.014), all events were considered not to be related to the treatment per the independent data safety monitoring board unblinded for the treatment assignment (table 2).
Serious adverse events during study follow-up
Discussion
In this interventional, proof-of-mechanism study, we show that a single infusion with rituximab is well tolerated and leads to a 12-month delay in the occurrence of clinical arthritis at the moment when 25% of the subjects had developed arthritis, when compared with placebo. The background risk of arthritis development of 40%, which is comparable to earlier reports of observational studies,3 10 was decreased by 55% at 12 months follow-up after treatment.
RA is one of the most common chronic immune-mediated inflammatory diseases with a significant impact on the individual patient as well as society. Current treatment options are still not sufficiently effective as most patients do not achieve disease remission, which is the treatment goal.24 25 Early initiation of treatment in patients diagnosed with RA increases the chance of better radiographic outcome and reaching long-term remission, a phenomenon referred to as the ‘therapeutic window of opportunity’. We are now capable of identifying individuals during an even earlier stage, when they are at risk of developing seropositive RA26 but before the onset of arthritis. This makes it possible to study whether there is a ‘preventive window of opportunity’.
While a previous, preventive intervention using dexamethasone has been proven unsuccessful,27 the results of our current study show that a single infusion of rituximab may alter the disease process, although temporarily. There are several possible mechanisms by which B lineage cells may contribute to the disease process, including antigen presentation, activation of T cells by providing costimulatory signals, production of proinflammatory cytokines and production of autoantibodies including RF and ACPA.28 Immune complexes containing RF or ACPA may directly activate macrophages, resulting in increased production of cytokines and chemokines like tumour necrosis factor and CXCL8 that are associated with the manifestations of clinical signs and symptoms.29 30
When the study was designed, we set to explore whether a single infusion of rituximab could reduce the risk of developing RA from the expected 40% to 10% in the studied population, a goal that was set to gratify the introduction of the treatment for this population, which was not achieved as the treatment resulted in markedly delayed onset of disease rather than cure. This result might be explained by persistence of autoreactive B-cell clones in the tissues and subsequent repopulation over time. It is tempting to speculate that repeated treatment, perhaps with a single infusion of rituximab once a year, might be sufficient to control B-cell numbers and prevent clinically manifest disease in a population at high risk of developing RA, whereas a more sophisticated approach would be to specifically target the autoreactive B cells that drive autoimmunity during the preclinical stage of the disease.5 If relevant autoantigens can be identified, one could envisage the development of therapies that target specifically autoreactive B cells in peripheral blood as well as in tissue niches such as lymph nodes and bone marrow, for example, by using autoantigen-based chimeric immunoreceptors that can direct T cells to kill autoreactive B lymphocytes through the specificity of the BCR.31 Alternatively, to achieve long-term prevention, polarised proinflammatory innate immune cells might need to be targeted in combination with B-cell depletion to stop reinitiation of the B-cell response, analogous to what has been proposed for the secondary prevention of type 1 diabetes in autoantibody positive subjects without clinically manifest diabetes.32 Clearly, further studies are needed to prove these concepts aimed at secondary prevention of RA; the current study supports the rationale for such future research. Other studies aimed at prevention of RA by changes of lifestyle-related risk factors, use of DMARDs and statins, or targeted therapies other than rituximab are currently under way.33
This study was subject to certain limitations. The biomarker analysis was exploratory in nature, and the relatively small sample size is a limitation. However, the results provide several interesting hypothesis-generating observations based on the changes in B-cell populations and B-cell products in relationship to treatment effects and development of arthritis over time. The results presented here are clearly consistent with the critical role of B cells in the pathogenesis of RA during the earliest stages of the disease and support future studies aimed at secondary prevention of RA, including by the use of targeted treatments.
Supplemental material
![[annrheumdis-2017-212763-supp2.jpg]](https://ard.bmj.com/content/annrheumdis/early/2018/12/17/annrheumdis-2017-212763/DC2/embed/inline-supplementary-material-2.jpg?download=true){kind=link}
Supplemental material
![[annrheumdis-2017-212763-supp3.jpg]](https://ard.bmj.com/content/annrheumdis/early/2018/12/17/annrheumdis-2017-212763/DC3/embed/inline-supplementary-material-3.jpg?download=true){kind=link}
Supplemental material
![[annrheumdis-2017-212763-supp4.jpg]](https://ard.bmj.com/content/annrheumdis/early/2018/12/17/annrheumdis-2017-212763/DC4/embed/inline-supplementary-material-4.jpg?download=true){kind=link}
Supplemental material
![[annrheumdis-2017-212763-supp5.jpg]](https://ard.bmj.com/content/annrheumdis/early/2018/12/17/annrheumdis-2017-212763/DC5/embed/inline-supplementary-material-5.jpg?download=true){kind=link}
Acknowledgments
We thank all the participants in this study, all participating institutions and their staff for their contribution to this study, including research physicians, research and specialised nurses. In addition, we thank Professor Dirkjan van Schaardenburg, Professor Tom Huizinga and Dr Elisabeth Brouwer for their valuable contribution in finding and encouraging eligible candidates to participate in this study. We thank Dr Marcel Dijkgraaf for biostatistical input in the set-up and initial analyses of the data, Dr Cristina Lebre and Tamara Ramwadhoebe for extensive support with the FACS analyses, Dr Daren Austin for statistical support, and Professor Lars Klareskog and Dr Linda Mathsson-Alm for measurement of ACPA antibodies. This manuscript is based on work previously presented at ACR 2016.
References
Footnotes
Handling editor Josef S Smolen
Presented at This work was previously presented at ACR 2016.
Correction notice This article has been corrected since it published Online First. The competing interest statement, abstract conlcusion and reference 20 have been updated.
Contributors PPT was the principal investigator, and was responsible for the study design, data interpretation and writing of the report. DMG and KIM contributed to the study design, data collection, data analysis, data interpretation and writing of the report. MS contributed to the data collection, data analysis, data interpretation and writing of the report. MWT, SWT, MdH, MJFSK, AvT, MJ, MH and NdV contributed to data collection and writing of the report. AHZ was responsible for data analysis, interpretation of the data and writing of the report.
Funding Funding was received from the Dutch Arthritis Foundation (grant number 11-1-407); Netherlands Organisation for Health Research and Development (ZonMw; grant number 200310003); European Union Seventh Framework Programme (project EuroTEAM; grant number FP7-HEALTHF2-2012-305549); IMI EU-funded project BeTheCure (grant number 115142).
Competing interests PPT is a former employee and DMG a current employee of GlaxoSmithKline, UK. GlaxoSmithKline was not involved in the design and/or execution of the study. NdV reports grants from AbbVie, Janssen Biologics, Ergomed Clinical Research, GlaxoSmithKline, Pfizer, Boehringer Ingelheim, and Roche, as well as personal fees from MSD, UCB, Janssen, non-financial support from Roche, personal fees and non-financial support from Pfizer, all outside the submitted work. In addition, NdV has a patent method for determining the risk of developing arthritis pending. MS reports a research grant from AstraZeneca (received in August 2015). AstraZeneca was not involved in this study.
Patient consent for publication Not required.
Ethics approval The Medical Ethics Research Committee of the Academic Medical Centre of the University of Amsterdam, The Netherlands.
Provenance and peer review Not commissioned; externally peer reviewed.