Article Text

Abstract
Objective Immune complexes (ICs) play a critical role in the pathology of autoimmune diseases. The aim of this study was to generate and characterise a first-in-class anti-FcγRIIA antibody (Ab) VIB9600 (previously known as MEDI9600) that blocks IgG immune complex-mediated cellular activation for clinical development.
Methods VIB9600 was humanised and optimised from the IV.3 Ab. Binding affinity and specificity were determined by Biacore and ELISA. Confocal microscopy, Flow Cytometry-based assays and binding competition assays were used to assess the mode of action of the antibody. In vitro cell-based assays were used to demonstrate suppression of IC-mediated inflammatory responses. In vivo target suppression and efficacy was demonstrated in FcγRIIA-transgenic mice. Single-dose pharmacokinetic (PK)/pharmacodynamic study multiple dose Good Laboratory Practice (GLP) toxicity studies were conducted in non-human primates.
Results We generated a humanised effector-deficient anti-FcγRIIA antibody (VIB9600) that potently blocks autoantibody and IC-mediated proinflammatory responses. VIB9600 suppresses FcγRIIA activation by blocking ligand engagement and by internalising FcγRIIA from the cell surface. VIB9600 inhibits IC-induced type I interferons from plasmacytoid dendritic cells (involved in SLE), antineutrophil cytoplasmic antibody (ANCA)-induced production of reactive oxygen species by neutrophils (involved in ANCA-associated vasculitis) and IC-induced tumour necrosis factor α and interleukin-6 production (involved in rheumatoid arthritis). In FcγRIIA transgenic mice, VIB9600 suppressed antiplatelet antibody-induced thrombocytopaenia, acute anti-GBM Ab-induced nephritis and anticollagen Ab-induced arthritis. VIB9600 also exhibited favourable PK and safety profiles in cynomolgus monkey studies.
Conclusions VIB9600 is a specific humanised antibody antagonist of FcγRIIA with null effector function that warrants further clinical development for the treatment of IC-mediated diseases.
- FcγRIIA
- antibody
- immune complex
- inflammation
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Immune complexes play a critical role in the pathology of a variety of autoimmune diseases. Immune complexes trigger FcγR-mediated inflammatory responses that are primarily driven through FcγRIIA. A variety of challenges have precluded the development of a therapeutic targeting this receptor.
What does this study add?
We generated a humanised effector-deficient anti-FcγRIIA antibody, VIB9600 which specifically binds to FcγRIIA and acts by both blocking ligand and internalising the receptor.
We demonstrated that VIB9600 suppresses immune complex-mediated activation of immune cells critical in the pathology of multiple autoimmune diseases both in vitro and in vivo.
How might this impact on clinical practice or future developments?
VIB9600 had a favourable pharmacokinetic and safety profiles in cynomolgus monkey studies and support its clinical development.
VIB9600 may provide a first-in-class treatment option towards immune complex-mediated autoimmune diseases.
Introduction
Autoantibodies directed against self-antigens may drive debilitating, organ specific or systemic manifestations which can be life-threatening.1 Spikes in autoantibody titres and changes in autoantibody profiles have been associated with autoimmune disease onset and flares in disease activity.2 Immune complexes (ICs) are found in the circulation and deposited in afflicted tissues and organs.3 4 Engagement of ICs with immune cells bearing Fc receptors can trigger cell recruitment and activation, localised inflammation, adaptive immunity and tissue pathology.4 5 Despite improved clinical management and the development of disease-modifying drugs, current therapies predominately target individual inflammatory pathways and many autoimmune patients fail to achieve remission.6 Available treatments including those targeting B cells only have modest effects on autoantibody titres, and it remains unclear if they have any impact on IC deposition.7 Consequently, interventions that block the engagement of ICs with FcγRs, antagonise activating FcγRs, or agonise inhibitory receptors, have been pursued for their therapeutic potential.8–10 To date, however, no therapies directly targeting FcγRs have been successfully developed.
In humans, the receptors for the Fc region of IgG (FcγRs) include the activating receptors FcγRI, FcγRIIA, FcγRIIIA and FcγRIIIB and the inhibitory receptor FcγRIIB.8 The activating receptor FcγRIIA has low affinity (KD ~10−6 M) for monomeric IgG,11 12 but the increased avidity afforded by the higher valency of aggregated IgG ICs permit FcγRIIA binding, clustering and signalling.11 There are two common human variants of FcγRIIA, 131 H and 131R, which exhibit differential binding toward IgG subclasses.13 Cross-linking FcγRIIA and phosphorylation of its immunoreceptor tyrosine-based activation motif triggers a signalling cascade that leads to multiple functional responses.5
FcγRIIA is an attractive target for therapeutic intervention as it is expressed on multiple immune cells that trigger pathological inflammatory responses.14 15 FcγRIIA has been specifically implicated in systemic lupus erythematosus (SLE), where its expression on plasmacytoid dendritic cells (pDC) drives IC-mediated type I interferon (IFN) production16 17 and its presence on neutrophils promotes lupus nephritis following the passive transfer of human SLE sera.18 In ANCA-associated vasculitis (AAV), autoantibodies targeting antigens exposed on neutrophils trigger FcγRIIA-dependent activation and tissue injury.19–21 ICs precipitated from rheumatoid arthritis (RA) may also trigger Fc-dependent induction of proinflammatory cytokines by monocytes.22
There are several challenges that need to be circumvented to facilitate the development of a therapeutic targeting FcγRIIA. With respect to specificity, it is noteworthy, that the extracellular region of FcγRIIA shares 94% identity with the inhibitory receptor FcγRIIB,23 so exposed epitopes that distinguish these receptors are limited. The two FcγRIIA 131 H and 131R variants are located in the ligand binding site,13 24 so ligand-blocking antibodies would need to be high affinity and preferably bind both allelic variants to be effective in the presence of high serum concentrations of IgG. It is also important that the therapeutic does not agonise FcγRIIA, trigger Fc-mediated hypersensitivity or induce effector functions such as complement-dependent cytotoxicity (CDC) or antibody (Ab)-dependent cell-mediated cytotoxicity (ADCC).11
The aim of this study was to develop an antibody targeting FcγRIIA suitable for the treatment of patients with IC-mediated disease. To that end, we generated VIB9600, a humanised, optimised, effector-deficient anti-FcγRIIA-specific antibody. We demonstrate that VIB9600 suppresses IC-mediated activation of immune cells critical in the pathology of multiple autoimmune diseases, and the pharmacology and safety data generated in non-human primate studies support its clinical development.
Results
Generation of VIB9600, a humanised effector-null FcγRIIA-specific antibody with dual mechanism of action
IV.3 is a well characterised murine IgG2b mAb specific for FcγRIIA.25 To generate a humanised antibody suitable for clinical development, the IV.3 complementary determing region (CDR) regions of the heavy and light chains were initially grafted on to the closest human germline variable heavy chain (Vh) and variable kappa chain (Vk) genes to generate Cam IV.3. Cam IV.3 was then optimised by screening amino acid substitutions in an epitope competition assay (with IV.3) to identify variants with improved binding. The sequences of the optimised, humanised antibody, VIB9600 and its parents IV.3 and Cam IV.3 are shown in online supplementary figure 1. VIB9600 has significant improvement in binding to FcγRIIA 131 H and 131R compared with Cam IV.3, and a modest improvement compared with IV.3, as assessed by epitope competition (figure 1A) and KD affinity measurements (table 1), while retaining the specificity for FcγRIIA (figure 1B).
Supplemental material

VIB9600 specifically binds FcγRIIA, competes with IgG for binding to FcγRIIA, causes receptor internalisation but fails to induce effector mechanisms. (A) Epitope competition data with IV.3 (mouse IgG 2b Ab), CamIV3 (humanised framework regions with IV.3 CDRs) and VIB9600 (humanised and optimised IV.3) on both human FcγRIIA 131 H (left) and FcγRIIA 131R (right). Representative data from two independent experiments are shown. (B) VIB9600 binding to human FcγRs in an ELISA-based binding assay. Plots represent the mean±SD. A representative plot of two independent experiments is shown. (C) In ADCC and CDC assays, the effects of VIB9600 were compared with wild type control 9600 IgG1 and isotype control IgG (R347-Tm) as indicated. in the ADCC assay, primary NK cells (effectors) were incubated with adherent FcγRIIA-expressing HEK-293 cells (targets) for 5 hour, and % cytotoxicity was determined. For CDC assays, baby rabbit complement was incubated with adherent FcγRIIA-expressing HEK-293 cells (targets), and % cytotoxicity was determined after 1 hour. Plots represent the mean±SD. Representative plots of three independent experiments are shown. (D) Binding of VIB9600 and control Ab (R347-Tm) to human FcγRIIA-expressing neutrophils in the presence and absence of 10 mg/mL IVIg (as indicated) was determined by flow cytometry (M.F.I). Representative data from two independent experiments. (E) Human monocytes were stained with CD14-Alexa 488 (green) and VIB9600-Alex 647 (red), and internalisation of FcγRIIA on human monocytes was visualised by confocal microscopy at time 0 and after culturing at room temperature for 1 hour. A representative image of three independent experiments is shown. (F) Available cell surface FcγRIIA on human monocytes and neutrophils in whole blood from healthy donors with either a 131 H/H or 131 R/R genotype (as indicated) was examined following a 2-hour incubation with VIB9600 or control Ab (R347-TM) by flow cytometry (M.F.I). (G) Similarly, cell surface FcγRIIA on cynomolgus monkey monocytes and neutrophils in whole blood was examined following a 12-hour incubation with VIB9600 or control Ab (R347-TM) by flow cytometry (M.F.I). Representative data from three independent humans and cynomolgus monkey experiments are shown. Ab, antibody; ADCC, antibody-dependent cell-mediated cytotoxicity; CDC, complement-dependent cytotoxicity; TM, triple mutation.
Binding affinity of IV.3 and VIB9600 Fabs to human FcγRIIA
A triple mutation (TM) (L234F/L235E/P331S) incorporated in the heavy chain constant region of VIB9600 was included to reduce Fc-mediated effector functions.26 To verify that the TM prevents Fc-mediated effector functions, VIB9600 and a variant incorporating an identical Fab with wild-type IgG Fc (9600 IgG1) were examined in ADCC and CDC assays. No notable ADCC or CDC was detected with VIB9600, whereas the variant with a wild-type Fc-induced cytotoxicity of FcγRIIA-expressing HEK cells in these assays (figure 1C), demonstrating that the TM renders VIB9600 effector null.
Next, we determined if VIB9600 has a competitive or non-competitive mode of action, by assessing the binding of VIB9600 to FcγRIIA-expressing neutrophils in the presence and absence of ligand. The presence of 10 mg/mL IVIG (pooled IgG) reduced the binding of VIB9600 to neutrophils from an EC50 of 0.03 nM to 3.35 nM (figure 1D). This competition for binding between the antibody and ligand indicates that the antibody has a ligand-blocking mechanism of action.
We next sought to determine if engagement of VIB9600 altered cell surface expression of FcγRIIA. Confocal microscopy indicated that incubation of VIB9600 with monocytes (1 hour at 37°C) resulted in the internalisation of FcγRIIA, whereas another antigen, CD14, remained expressed on the cell surface (figure 1E). VIB9600-mediated internalisation of FcγRIIA was verified using a FACS-based whole blood assay on human monocytes and neutrophils from donors with either a 131 H/H or a 131 R/R genotype (figure 1F). To justify, cynomolgus monkey as a relevant pharmacology and toxicology species, we also demonstrated that VIB9600 also reduced cell surface bound FcγRIIA on monocytes and neutrophils from cynomolgus monkey whole blood (figure 1G). Together these data demonstrate that VIB9600, has two significant modes of action: it blocks ligand and reduces the cell surface expression of FcγRIIA available for ligand engagement.
VIB9600 blocks autoantibody/IC-mediated inflammatory responses
Next, we assessed the capacity of VIB9600 to block IC-mediated inflammatory responses driven by different cell types relevant to autoimmune diseases. DNA/RNA associated ICs trigger pDC to produce the type I IFNs which are implicated in the pathogenesis of SLE.16 Using peripheral blood mononuclear cells (PBMCs) as a source of pDC, VIB9600 potently inhibited the induction of type I IFN induced by ribonucleoprotein IC (RNP-IC) from healthy donors with either a 131 H/H or a 131 R/R genotype (figure 2A). IC can also trigger monocytes to produce TNFα and IL-6, key cytokines involved in the pathogenesis of RA.22 Compared with control antibody, VIB9600 inhibited the IC induction of IL-6 and TNFα in whole blood by approximately 60% and 80%, respectively (figure 2B). In AAV, autoantibodies against cytoplasmic antigens such as MPO and PR3 lead to neutrophil activation, and the induction of reactive oxygen species (ROS) which induces tissue pathology.19 Pretreatment of neutrophils with VIB9600 inhibited anti- myeloperoxidase (MPO) and anti-proteinase 3 (PR3) antibody-induced superoxide production as determined using either ferri-cytochrome c reduction assay (figure 2C) or oxidation of dihydrorhodamine 123 (DHR123) (figure 2D). VIB9600 also blocked ROS production from neutrophils stimulated with IgG-purified AAV patient’s sera seropositive for either anti-PR3 or anti-MPO antibodies (figure 2E). Taken together these data demonstrate that VIB9600 can inhibit autoantibody and IC-mediated activation of inflammatory processes associated with autoimmune diseases.

VIB9600 blocks autoantibody/IC-mediated inflammatory responses. (A) VIB9600 inhibited RNP-IC induced IFNα protein produced from human PBMC. Representative dose response curve from three independent experiments with human 131 H/H and 131 R/R donors are presented. (B) VIB9600 (30 µg/mL) inhibited Ig-IC-induced TNF-α and IL-6 protein in whole blood. Mean±SD percentage inhibition relative to no antibody are presented. *P<0.05, paired Student t-test. (C) VIB9600 inhibition of ANCA-induced neutrophil superoxide production measured by ferri-cytochrome C reduction assay. Human neutrophils were primed with 2 ng/mL TNF-α, with or without VIB9600 and stimulated with anti-MPO (left) or anti-PR3 Ab (right): data represent the mean±SD (n=4 replicates) of ΔOD550–490 values. Representative plots from two independent experiments are presented. (D–E) Effect of VIB9600 blockage on ANCA-induced neutrophil activation in a DHR123 assay. (D) Experiment showing the oxidative burst of neutrophils activation from TNF-α-primed human neutrophils stimulated with an anti-MPO antibody (left) or an anti-PR3 antibody (right) and treated with the indicated reagents. Oxidation of DHR123 was measured by flow cytometry, and the data show changes in M.F.I. Data were generated from three independent experiments. Error bars represent the mean±SD. (E) Left: experiment showing the oxidative burst from TNF-α-primed human neutrophils stimulated with IgG isolated from AAV anti-MPO-positive patient sera and IgG isolated from healthy volunteer (HV) sera with and without VIB9600. Right: same experiment with IgG isolated from AAV anti-PR3-positive patient sera. DHR123 oxidation was measured by flow cytometry; data show changes in M.F.I. Data were generated from three independent experiments. Error bars represent the mean±SD. *P<0.05, **p<0.01, ***p<0.001, paired Student t-test. AAV, ANCA-associated vasculitis; Ab, ANCA, antineutrophil cytoplasmic antibody; IL-6, interleukin-6; IC, immune complex; RNP-IC, ribonucleoprotein IC; Tm, triple mutation; TNF-α, tumour necrosis factor-α.
VIB9600 does not adversely impact neutrophil function or agonise FcγRIIA in vitro
Neutrophils play a critical role in host defense by sensing infection and tissue injury and initiating an acute inflammatory response.27 28 Therefore, it was important to determine if VIB9600 inadvertently activates neutrophils or otherwise impedes their function. Importantly, VIB9600 did not impact phorbol 12-myristate 13-acetate (PMA)-mediated ROS production (figure 3A), Pam3CysSerLys4 (TLR2)-induced CD11b upregulation (figure 3B) or IL-8 mediated neutrophil migration (figure 3C). Finally, we examined the impact of VIB9600 on antibody-dependent (anti-PsI mAb PsI0096) opsonophagic killing (OPK) of Pseudomonas aeruginosa, a clinically important antibiotic-resistant strain of bacteria.29 VIB9600 had a minimal impact on P. aeruginosa OPK, whereas blockade of FcγRIII significantly inhibited OPK activity (figure 3D). Together these data indicate that VIB9600 does not inadvertently impact neutrophil functions.

Blockade of FcγRIIA by VIB9600 has no adverse effects on neutrophil function and has no impact on protein expression in a whole blood proteomic assessment. (A) Effect of VIB9600 on PMA-induced reactive oxygen species production measured by ferri-cytochrome c reduction assay. ΔOD550-490 values obtained from TNF-α-primed human neutrophils stimulated with PBS, PMA or VIB9600+PMA. A representative plot of two independent experiments is shown. Error bars represent the mean±SD from one experiment. (n=4 replicates). (B) Effect of VIB9600 on neutrophil activation. M.F.I values for the cell surface expression of CD11b which is the indication of neutrophils activation from human neutrophils treated with the indicated reagents. Error bars represent the mean±SD from three independent experiments. (C) Effect of VIB9600 on neutrophil migration. Migration index values (the ratio of the number of cells that migrated in response to the reagent versus the number that migrated without it) obtained from human neutrophils treated with the indicated reagents. Error bars represent the mean±SD from three independent experiments. (D) Effect of VIB9600 on antibody-mediated phagocytosis. representative data of three independent opsonophagocytic killing assays. VIB9600, anti-FcγRIIB mAb or anti-FcγRIII mAb was preincubated with neutrophils. Dilutions of the anti-Psl antibody Psl0096, complement and luminescent bacteria were then added to each well and incubated for 120 min at 37°C. Relative luciferase units (RLU) were measured. The percent killing of Pseudomonas aeruginosa was calculated using the following formula: % Killing=100−([RLU experimental wells/RLU control wells]×100). Error bars represent the mean±SD. (E) Effect of VIB9600 in whole blood. There are five individual donors (each column) in each treatment group. Data were z-score transformed, and heatmaps were generated in R using the heatmap.2 function of the gplots package. Samples were clustered by condition, although the protein clustering structure was unsupervised. Ab, RNP-IC, ribonucleoprotien-immune complex.
Crosslinking FcγRs has the potential to stimulate the secretion of inflammatory cytokines30 or induce immune hypersensitivity.31 To assess the agonistic potential of VIB9600, whole blood was treated with 30 µg/mL of VIB9600, or ICs (positive control) for 16 hours at 37°C, and changes in secreted protein expression were examined. Cross-linking FcγRs with IgG-IC or RNP-IC-induced profound changes in the protein levels, however, there was no discernable difference between the protein profiles of untreated and VIB9600-treated samples (online supplementary table 1 and figure 3E). These data indicate that VIB9600 does not exhibit agonistic activity.
VIB9600 suppresses antibody-mediated pathology in FcγRIIA transgenic mice
Since FcγRIIA does not exist in rodents, we assessed the pharmacology of VIB9600 in FcγRIIA transgenic mice. First, we demonstrated that VIB9600 transiently reduced FcγRIIA expression on platelets and neutrophils in a dose-dependent manner over a 4-day period (figure 4A). With 10 mg/kg of VIB9600, no free surface FcγRIIA was detected on platelets and neutrophils through 48 hours (figure 4A), which is consistent with the presence of circulating VIB9600 at that time (figure 4B). In FcγRIIA transgenic models of autoimmunity, VIB9600 inhibited antiplatelet-induced thrombocytopaenia, neutrophil infiltration in acute antiglomerular basement membrane (GBM)-induced nephritis model and anticollagen Ab-induced arthritis (figure 4C–E). These data demonstrate a direct relationship between VIB9600 levels, FcγRIIA target engagement and efficacy in IgG antibody-mediated autoimmune disease models.
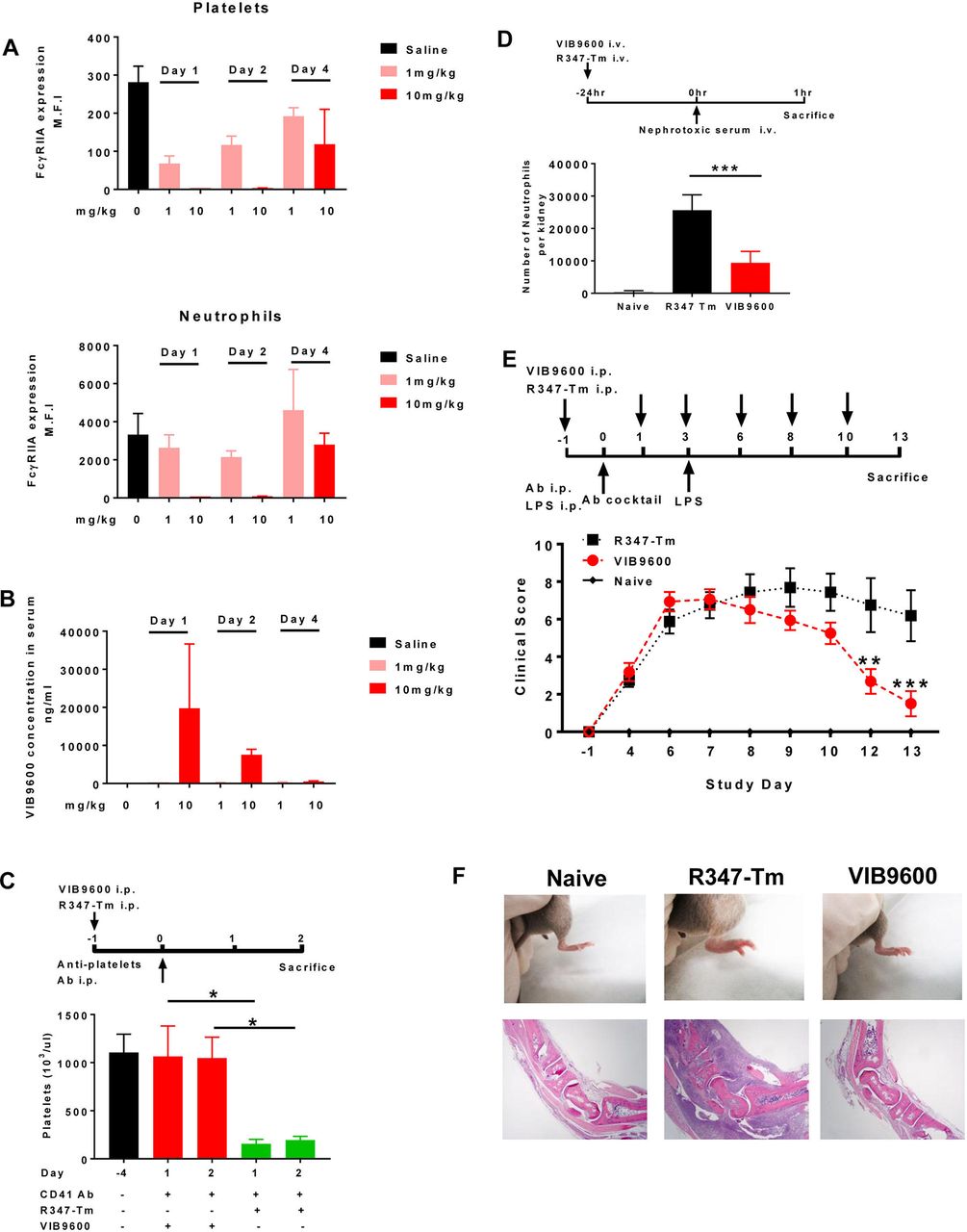
VIB9600 suppressed the FcγRIIA and antibody-mediated pathology in FcγRIIA transgenic mice. (A+B) The pharmacology of VIB9600 was assessed in FcγRIIA transgenic mice. Mice were treated with 1 mg/kg or 10 mg/kg VIB9600 by intraperitoneal at day 0, blood was collected at day 1, day 2 and day 4 postinjection. (A) Free FcγRIIA on platelets and neutrophils in FcγRIIA transgenic mice was measured by flow cytometry at 24, 48 and 96 hours after a single 1 mg/kg or 10 mg/kg intraperitoneal dose of VIB9600. (B) Serum concentrations of VIB9600 were measured by human IgG ELISA at 24, 48 and 96 hours after 1 mg/kg or 10 mg/kg intraperitoneal dose of VIB9600. (C) Effect of VIB9600 in FcγRIIA transgenic model of antibody-induced thrombocytopaenia. VIB9600 or control Ab (R347-Tm) was injected intraperitoneal at 10 mg/kg 24 hours (day 1) before thrombocytopaenia was induced with 2 µg rat antimouse CD41Ab delivered intraperitoneal at day 0, platelets numbers were determined at day 4 (baseline) and following induction of thrombocytopaenia (day 1 and day 2). A representative plot of two independent experiments is shown. Error bars represent the mean±SD from one experiment. (n=3 mice/group). *P<0.05 by unpaired Student’s t-test. (D) Effect of VIB9600 in FcγRIIA-mediated neutrophil infiltration in an acute model of anti-glomerular basement membrane (aGBM) nephritis. Transgenic mice with FcγRIIA expression selectively in neutrophils of mice lacking endogenous murine receptors were given 20 mg/kg of VIB9600 or isotype control by intravenous injection 24 hours before intravenous injection of nephrotoxic serum (see timeline). Mice were euthanised, and kidneys and blood were collected for FACS analysis. Infiltrating renal neutrophils per kidney were quantitated by flow cytometry. naïve mice with no treatment were also euthanised and analysed. Bar graph represents mean±SD for nine animals in each treated group and four animals in the naïve, untreated group. ***P<0.001 by unpaired Student’s t-test. (E–F) Effect of VIB9600 in FcγRIIA transgenic model of anticollagen Ab-induced arthritis. (E) VIB9600 or control Ab (R347-Tm) was injected intraperitoneal at 20 mg/mL at day -1,1,3,6,8 and 10, arthritis was induced with intraperitoneal. Delivery of 2 mg anticollagen Ab cocktail at day 0 and 10 μg lipopolysaccharide (LPS) at day 3. Arthritis was evaluated by clinical score at indicated timepoint. Error bars represent the mean±SD (n=8 mice/group). Two-way analysis of variance analysis, **P<0.01, ***p<0.001. (F) Top panel: representative image of hind paws at day 13 after the initial injection of anticollagen Ab cocktail. (F) Bottom panel: photomicrography analysis of H&E stained tissue sections from tarsal joint obtained from representative mice. 4X obj. Ab, antibody.
Pharmacokinetic and exploratory pharmacodynamic and GLP toxicity studies of VIB9600 in cynomolgus monkeys
To establish the pharmacokinetic/pharmacodynamic (PK/PD) characteristics of VIB9600, a single ascending dose study was conducted in cynomolgus monkeys (online supplementary table 2). The non-compartmental analysis indicated that the terminal half-lives for VIB9600 were 1.08–1.2 days in the 1 mg/kg group, increasing to 2.84–4.3 days in the 100 mg/kg group (figure 5A). The tendency for half-life to increase with dose is consistent with target-mediated elimination of the antibody. The PD characteristics of VIB9600 were measured by flow cytometry to assess free FcγRIIA on the surface of cells. A single-dose of VIB9600 induced a dose responsive and completely reversible reduction of FcγRIIA on monocytes (CD45+, CD14+) and granulocytes (side scatter/CD45+) (figure 5B,C). Notably, the rapid suppression and slower recovery of FcγRIIA expression mirrored the PK profile of VIB9600. These data indicate a strong and direct relationship between PK exposure and PD response in the form of FcγRIIA expression (figure 5A–C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Single-dose pharmacokinetic and exploratory pharmacodynamic study of VIB9600 in cynomolgus monkeys. Male monkeys were given VIB9600 once at 1, 10 or 100 mg/kg in a volume of 2 mL/kg via intravenous injection. (A) Serum concentration of VIB9600 in cynomolgus monkeys at various time points after a single dose was determined by ELISA. (B) and (C) FcγRIIA levels on monocytes and neutrophils were determined at different time points by flow cytometry. The FcγRIIA levels (average percentage) relative to the mean predose levels are shown.
To assess the safety of VIB9600, we conducted a 3-month subcutaneous and intravenous GLP toxicity study (13 weekly doses up to 100 mg/kg and an 8 week follow-up, online supplementary table 2). This GLP safety study did not result in any changes in body weight or platelet counts, and FcγRIIA levels returned to predose levels by the end of the study period (online supplementary figure 2). In addition, no adverse effects on other haematology, clinical chemistry, organ weight or histopathology were noted (data not shown).
Discussion
IC and autoantibodies targeting specific cells and tissues can engage Fc-bearing immune cells and drive leucocyte recruitment and local tissue pathology. We proposed that suppressing IC-mediated cellular activation by inhibiting FcγRIIA would provide an alternate therapeutic approach for the treatment of autoimmune conditions. Herein, we generated VIB9600, a humanised, optimised, anti-FcγRIIA antibody with null effector function that potently inhibits IC-mediated responses from multiple cell types that are believed to play a critical role in autoimmune diseases. In vivo studies demonstrate a direct relationship between VIB9600 PK and target engagement, and safety assessments in vitro and in non-human primates support its clinical development.
To the best of our knowledge, no molecules directly targeting FcγRIIA have entered clinical development. Targeted therapeutic efforts that interfere with IC-mediated activation of FcγR have recently emerged. Recombinant FcγRIIB (SM101), which has the potential to sequester ICs, showed some efficacy in phase II clinical trials in immune thrombocytopenic purpura (ITP) and SLE.32 In addition, an anti-FcγRIIB Ab (SM201) has been designed to recruit FcγRIIB to trigger and enhance inhibitory signals.32 33 M230, a recombinant trivalent-IgG-Fc engineered for increased binding to FcγRs, was also reported to block Fc-mediated IC activation in preclinical studies.34 Potentially, M230 could block interactions with all FcγR, so its pharmacology and impact on the clearance of IC and opsonised pathogens will be of interest. FcγRs mediate phagocytosis of large IgG-coated particles and pinocytosis of soluble IgG ICs.11 35 36 It has been shown that neutrophil FcγRIIIB plays a dominant role in the homeostatic clearance of soluble ICs,36 but it remains uncertain what impact the complete blockade of FcγRs will have on the clearance of ICs. Since VIB9600 only inhibits FcγRIIA, it is predicted that other FcγRs and the complement system should adequately remove complexes from the circulation.
There remains a critical need for safe and efficacious drugs in autoimmune disease. In SLE, the only approved drug in the last 60 years, belimumab, an antibody antagonist of the B cell growth and differentiation factor BLYS, has only a modest response compared with standard of care.37 38 We demonstrate that VIB9600 potently inhibits the induction of type I IFNs by pDCs stimulated with RNA-containing IgG complexes. A type I IFN gene signature is prominent in ~75% of patients with SLE, and a recent successful double-blinded phase II clinical trial targeting interferon-alpha/beta receptor alpha chain (IFNAR1) demonstrates the importance of this pathway.39 40 Besides blocking IC-mediated induction of type I IFNs by pDC, targeting FcγRIIA with VIB9600 will also inhibit IC-mediated induction of other inflammatory mediates from antigen-presenting cells, granulocytes and platelets.41 IC-mediated activation of FcγRIIA on neutrophils has also been reported to trigger formation of neutrophil extracellular traps and promote autoimmunity by providing an immunogenic source of autoantigens.36 Therefore, it is tempting to speculate that VIB9600 could provide a greater benefit in SLE than current therapeutics.
In AAV, antibodies targeting cytoplasmic antigens MPO and PR3 exposed on the surface of neutrophils can trigger endothelial adhesion, degranulation and the release of proteolytic enzymes and ROS which drive vascular injury.19–21 VIB9600 potently suppressed ANCA-induced neutrophil activation and the production of ROS. Importantly, despite targeting a neutrophil cell surface receptor (FcγRIIA), VIB9600, did not inappropriately activate neutrophils nor block other neutrophil functions. Current treatments for AAV include cyclophosphamide or rituximab in combination with steroids. High-dose steroids drive significant morbidity and repeated cycles of cyclophosphamide are contraindicated. Rituximab induces remission in about 60% of treated patients,42 but it remains uncertain to what extent this effect is driven by steroids, and the relapse rate during the first year after induction remains high. Interestingly, data from rituximab in ANCA-associated vasculitis trial indicated differential response rates for the different FcγRIIA 131 H/R alleles.43 This implies that FcγRIIA plays a critical role for in the disease pathogenesis, and importantly VIB9600 binds and inhibits both allelic variants similarly. If blockade and internalisation of FcγRIIA translate to a more rapid and sustained clinical response particularly if the use of steroids could be reduced, VIB9600 may provide clinical advantages.
The demonstration that VIB9600 can inhibit IC-mediated induction of TNFα and IL-6 among other proinflammatory molecules would suggest that VIB9600 may provide an alternative treatment option for RA. Treatments targeting the TNFα or IL-6 pathways have been approved for RA highlighting the importance of these cytokines in the pathology of the disease.44 45 Although it is unclear to what extent the production of these cytokines is driven by FcγRIIA activation in RA, it is tempting to speculate that targeting FcγRIIA could be a more beneficial therapeutic approach than targeting downstream cytokines individually.
Besides the compelling evidence that VIB9600 can block key IC-mediated inflammatory responses in multiple human cells, we demonstrate that VIB9600 blocks antiplatelet-mediated thrombocytopaenia and anticollagen Ab-induced arthritis in FcγRIIA transgenic mice. This is consistent with previous in vivo studies that established the balance of activating and inhibitory FcγRs is important in triggering autoimmune disease,46 experimental models of ITP, RA and haemolytic anaemia,47–49 VIB9600 also reduced neutrophil accumulation in anti-GBM nephritis, which is consistent with the role of neutrophil FcγRIIA in the pathogenesis of glomerulonephritis50 However, the individual expression profiles and functions of FcγR differ so significantly between humans and mice, that the role of FcγRIIA in human autoimmune diseases may only be faithfully assessed in human clinical trials.
Taken together, there remains a significant unmet need in SLE, AAV, RA and other IC-mediated and antibody-mediated autoimmune conditions for safe, fast-acting efficacious drugs that have durable effects and can significantly reduce corticosteroid usage. There is strong rationale for targeting FcγRIIA in these diseases, and VIB9600 may provide a first-in-class treatment option. VIB9600 potently inhibits FcγRIIA-mediated responses, and preclinical pharmacology and safety assessments support its clinical development to assess its efficacy in autoimmune diseases.
Methods
Binding affinity and specificity were determined by Biacore and ELISA. Confocal microscopy, FACS-based assays and binding competition assays were used to assess the antibody mode-of-action. ADCC and CDC assays were performed using human embryo kidney (HEK)-293 stably transfected FcγRIIA cells. IC-induced ROS was measured using ferri-cytochrome C reduction and oxidation of DHR123 assays. Pam3CSK4 (TLR2)-induced neutrophil activation was assessed by CD11b upregulation. Cell migration was measured using a 96-well Chemo TX system. OPK of P. aeruginosa with PsI antibody PsI0096 was assessed with luminescent P. aeruginosa cells. FcγRIIA cross-linking was assessed in whole blood from heparin tubes, and following stimulation with VIB9600 or IC, protein analytes were measured using myriad multianalyte profiling technology platform. Target suppression was examined in FcγRIIA transgenic (B6: SJL-Tg (FcγRIIA)11Mkz/J) mice from Jackson Laboratory, and cynomolgus monkeys using a FACS-based receptor occupancy assay. In vivo efficacy was assessed using anti-CD41 Ab immune-mediated thrombocytopaenia model, acute anti-GBM nephritis model and anticollagen Ab-induced arthritis model in FcγRIIA transgenic mice. Further details of these assays are available in supplementary materials.
Statistical analysis
The statistical significance of differences between two groups was analysed using Student’s t-test. Statistical significance was ascribed to the data when p<0.05.
Acknowledgments
We thank Dr. Tomas Mustelin for helpful discussion and Medimmune’s laboratory animal resource’s excellent technical assistance.
References
Footnotes
Handling editor Josef S Smolen
Correction notice This article has been corrected since it published online first. The author name Martin J Borrok has been removed as this is a redundant name in the authorship.
Contributors BC, KV, GS: developed the concept and wrote the paper. BC, ST, HS, SW, LV, BK, SK, DR, BN, EG, MH, SE, AD, MB, XX, WZ, JC, HK, CM, YW, JC, WZ, HK, KO, TM, YZ: generated and interpreted data. DH, JG, RK, RH: supervised project. All authors critically reviewed and edited the paper.
Funding This study was sponsored by MedImmune/AstraZeneca and Viela Bio.
Competing interests MedImmune employees hold stock in AstraZeneca. Shu Wang is the emploee of the Viela Bio. Viela Bio is the sole owner of VIB9600. Bing Yao (YaoB@vielabio.com) is the CEO of Viela Bio and VIB9600 is in clinical development.
Patient consent Not required.
Ethics approval Blood from healthy volunteers was obtained with informed consent under MedImmune, LLC’s blood donation program, and studies using human cells were performed in accordance with the Institutional Review Board guidelines. For animal studies, all procedures were performed in accordance with federal, state and institutional guidelines in an AAALAC-accredited facility and were approved by the MedImmune Institutional and The Brigham and Women’s Hospital Animal Care and Use Committee.
Provenance and peer review Not commissioned; externally peer reviewed.