Article Text

Abstract
Objectives To evaluate the long-term efficacy and safety of canakinumab in patients with active systemic juvenile idiopathic arthritis (JIA).
Methods Patients (2–19 years) entered two phase III studies and continued in the long-term extension (LTE) study. Efficacy assessments were performed every 3 months, including adapted JIA American College of Rheumatology (aJIA-ACR) criteria, Juvenile Arthritis Disease Activity Score (JADAS) and ACR clinical remission on medication criteria (CRACR). Efficacy analyses are reported as per the intent-to-treat population.
Results 144 of the 177 patients (81%) enrolled in the core study entered the LTE. Overall, 75 patients (42%) completed and 102 (58%) discontinued mainly for inefficacy (63/102, 62%), with higher discontinuation rates noted in the late responders group (n=25/31, 81%) versus early responders (n=11/38, 29%). At 2 years, aJIA-ACR 50/70/90 response rates were 62%, 61% and 54%, respectively. CRACR was achieved by 20% of patients at month 6; 32% at 2 years. A JADAS low disease activity score was achieved by 49% of patients at 2 years. Efficacy results were maintained up to 5 years. Of the 128/177 (72.3%) patients on glucocorticoids, 20 (15.6%) discontinued and 28 (22%) tapered to 0.150 mg/kg/day. Seven patients discontinued canakinumab due to CR. There were 13 macrophage activation syndrome (three previously reported) and no additional deaths (three previously reported). No new safety findings were observed.
Conclusion Response to canakinumab treatment was sustained and associated with substantial glucocorticoid dose reduction or discontinuation and a relatively low retention-on-treatment rate. No new safety findings were observed on long-term use of canakinumab.
Trial registration numbers NCT00886769, NCT00889863, NCT00426218 and NCT00891046.
- canakinumab
- clinical trial
- interleukin-1β
- long-term extension
- systemic juvenile idiopathic arthritis.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Key messages
What is already known about this subject?
The key role of IL1 in the pathogenesis of sJIA and the therapeutic implications from its blocade.
Canakinumab, a fully human monoclonal Ab which selectively blocks IL1 beta in patients with sJIA, has proved its efficacy and safety during a phase II and phase III clinical program.
What does this study add?
The study provides with long term (up to 5 years) safety and drug survival data on a pooled population from canakinumab’s clinical program. Canakinumab’s effect on systemic features and joints proved to be maintained in the long term particularly in the early responders patients. For the first time early response has been shown to be linked to canakinumab’s long term survival rendering it as an easy identifiable clinical predictor factor of long term maintenace of remission/low disease activity.
How might this impact on clinical practice or future developments?
As long as achievement, as much as maintenance of remission or alternatively low disease activity constitute the ultimate therapeutic target in order to prevent future organ damages and disease related comorbidities, time to response will facilitate physicians in their decision making to keep or switch canakinumab to another treatment in a timely manner.
Introduction
Currently available therapies for systemic juvenile idiopathic arthritis (sJIA) include non-steroidal anti-inflammatory drugs, glucocorticoids, synthetic disease-modifying antirheumatic drugs (DMARD) and biologic DMARDs that inhibit primarily interleukin (IL)-6 and IL-11–7; tumour necrosis factor blockers and CTL4-Ig for sJIA without systemic features.8–13 Management of sJIA is aimed at achieving and maintaining clinical remission (CR). Minimising glucocorticoid exposure, ideally up to discontinuation, is also of foremost importance in an effort to prevent the inhibition of growth.14 15
IL-1 plays a key role in the pathogenesis of sJIA.16 Several reports have suggested that inhibition of IL-1 provides clinical benefit in sJIA.1 17 Canakinumab is a fully human monoclonal antibody that selectively binds to IL-1β, inactivating its downstream signalling cascade. Previous phase II–III trials have demonstrated the efficacy and safety of canakinumab in patients with sJIA.4 5
Here, we report the long-term efficacy and safety of canakinumab in patients with sJIA with active systemic features and arthritis at baseline, who were enrolled from the previously reported pivotal phase III studies,5 and followed for up to 5 years.
Methods
Study design
The study design of the two pivotal phase III trials has been previously reported.5 Briefly, in trial 1 of 1-month duration, a single canakinumab dose or placebo was administered. Patients from trial 1 could enter the two-part trial 2 where canakinumab-naïve patients and patients from a phase II trial4 were additionally enrolled. Trial 2 was a randomised withdrawal study,18 with an open-label lead in part I up to 32 weeks. Glucocorticoid tapering was permitted, and monitored by the Paediatric Rheumatology International Trials Organisation (PRINTO) and the Pediatric Rheumatology Collaborative Study Group (PRCSG) coordinating centres,19 based on disease activity level5 (achieved at least an American College of Rheumatology (ACR) 50 with no fever and C-reactive protein (CRP) <10 mg/L; further details in (online supplementary file 1). Part I of trial 2 was followed by a randomised, double-blind, placebo-controlled, event-driven withdrawal part II in which adapted JIA American College of Rheumatology (aJIA-ACR) 30 responders able to taper/discontinue glucocorticoids were randomised to receive placebo or continue canakinumab until the end of trial 2 or a flare of sJIA had occurred (part II; average total study duration of 29.5 weeks).20 Patients from trial 2 (figure 1) were allowed to enter the open-label, long-term extension (LTE) study where patients were planned to be followed for a minimum of 96 weeks, with further glucocorticoid tapering as per physician’s decision. Patients received canakinumab 4 mg/kg subcutaneously every 4 weeks (maximum dose: 300 mg); canakinumab dose was tapered in the LTE to 2 mg/kg every 4 weeks in patients who were glucocorticoid free as per physicians’ judgement.
Supplemental material

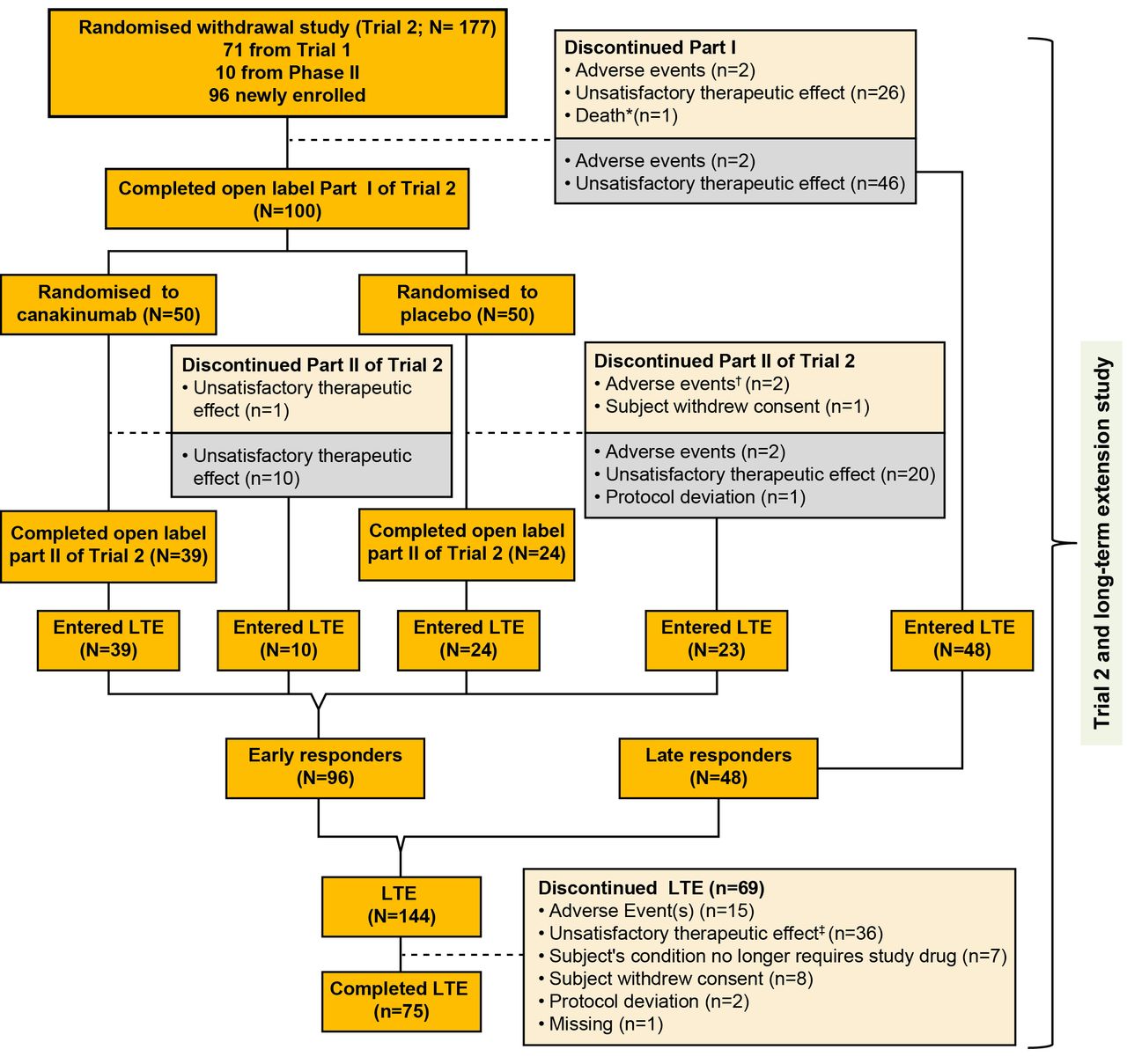
Flow chart with patient disposition. *One death occurred during part I; patient died due to MAS. †A patient in the placebo group died due to MAS 2 days after discontinuing the part II phase due to MAS. ‡One patient died from disease progression 3 months after discontinuation from the long-term extension phase due to unsatisfactory therapeutic effect. The grey box represents the patients who discontinued the part I or part II of trial 2 and entered the long-term extension study. Patients who entered the LTE are divided into two subgroups: (1) early responders, defined as patients who had successfully completed the glucocorticoid tapering in part I of trial 2 as per protocol and who were randomised to the withdrawal part; (2) late responders, defined as patients who moved directly from the open-label part of trial 2 and who failed to taper glucocorticoids in part I. LTE, long-term extension; MAS, macrophage activation syndrome.
Patients
Patients were followed up in the LTE study between 6 July 2009 and 5 December 2014 at 63 centres of PRINTO/PRCSG in 21 countries. Eligibility criteria for the phase III trials have been described previously.5 In brief, eligible children (2–19 years old) with confirmed sJIA as per the International League Against Rheumatism classification criteria, active systemic features of sJIA, at least two active joints, CRP level >30 mg/L (normal range: 0–10 mg/L) and being treated with a prednisone equivalent of ≤1.0 mg/kg/day were included. Major exclusion criteria included macrophage activation syndrome (MAS) within the last 6 months, active infections, malignancies and concurrent use of other biologics.
Assessments
Efficacy assessments were performed at least every 3 months to assess the levels of improvement using various composite validated measures: aJIA-ACR 50/70/90, based on the JIA core set variables,21–24 plus the absence of fever (defined as temperature ≤38°C in the preceding 7 days); clinically inactive disease (CID)/clinical remission on medication (CR), defined as at least 6 months of CID, which were evaluated by either the ACR criteria (CIDACR/CRACR)25 26 or by the Juvenile Arthritis Disease Activity Score 71-CRP (JADAS; CIDJADAS/CRJADAS).27 Disease activity was measured by JADAS score with the following cut-offs: CIDJADAS score ≤1; low disease activity (LDA) score ≤3.8; moderate disease activity score 3.9–10.5; and high disease activity (HDA) score >10.527–29; systemic features were reflected in the physician global evaluation of disease activity measured on a visual analogue scale.
Safety and tolerability of canakinumab were assessed in terms of adverse events (AE), serious AEs (SAE) and clinical and laboratory assessments from first injection until the last available observation. Serious infections, malignancies and cases of MAS were adjudicated by independent committees.30–33
A three-tiered approach was used to measure anti-canakinumab antibodies (anti-drug antibody (ADAs)), consisting of a screening, a confirmatory and a titration assay, respectively. Serum canakinumab concentrations (pharmacokinetics, PK) were determined to assess the relationships between canakinumab exposure and the immunogenicity data.34 35
Statistical analysis
The European League Against Rheumatism (EULAR) recommendation for reporting LTE studies36 and the Consolidated Standards of Reporting Trials statement37 38 were followed. Categorical variables were summarised by absolute frequencies and percentages, while continuous variables were summarised by median and lower and upper quartiles. The aJIA-ACR criteria used the starting day of canakinumab as baseline. Efficacy analyses were performed in two ways: (1) for primary analysis: in the intent-to-treat (ITT) population based on observed data with all discontinuations at different time points counted as missing (n=177; patients enrolled in trial 2) and (2) with missing data imputed using last observation carried forward (LOCF) (online supplementary appendix).39
In exploratory analysis, the hypothesis was tested that early responders achieve more substantial sJIA control as compared with late responders of canakinumab. For the purpose of this analysis, early responders were defined as patients who had successfully completed the glucocorticoid tapering in part I of trial 2 as per protocol and who were randomised to the withdrawal part; late responders were defined as patients who moved directly from the open-label part of trial 2 because they failed to taper glucocorticoids in part I. Mixed model analyses were used to quantify the difference in efficacy responses for early and late responders. Subgroup exploratory analyses were also considered by methotrexate (MTX) coadministration and prior use of biologics using the generalised mixed model framework. All mixed models were adjusted for repeated measures, and include a centre-related random effect and the following covariates: time in study, prior use of biologics, prior use of MTX, prior use of corticosteroid, number of joints with limitation of motion (LOM) and number of active joints.
AEs were summarised by presenting the number of events and percentage of patients and time-adjusted frequencies of exposure as per the primary system organ class based on the Medical Dictionary for Regulatory Activities (MedDRA, Version 17.1) preferred term.
Results
Baseline characteristics and patient disposition over time
Demographics and baseline disease characteristics of the 177 patients at baseline as well as the 144 patients rolling over to the LTE study are presented in table 1. At baseline, as previously reported, 99.4% of patients had HDA (median JADAS=32.5).
Demographics and clinical characteristics of patients at baseline (date of first canakinumab administration and at the time of entry into the LTE study)
Of the 177 patients (per ITT, denominator is 177 unless otherwise stated) enrolled in trial 2, a total of 144 (81%) continued in the LTE study (figure 1); 122 (69%) stayed for a maximum of 271 weeks. Overall, 75 (42%) patients completed the LTE study.
As shown in table 1, among the 144 patients who entered LTE, there were 96 (54.2%) early responders with a median JADAS of 1.85 denoting an LDA status (62% JADAS LDA and 47.9% CIDJADAS) at baseline of the LTE study, and 48 (27.1%) late responders with a median JADAS of 18.8 denoting HDA status (69% with JADAS HDA at baseline of the LTE). Compared with early responders, the late group had a higher frequency of prior use of biologics (79.2% vs 57.3%) and required higher glucocorticoid doses (0.36 mg/kg/day vs 0.17 mg/kg/day). Similar differences were noted at baseline (day of the first canakinumab administration) in the two groups.
A total of 102 (58%) patients discontinued canakinumab: 33 during trial 2 as previously reported5 and the remaining 69 during the LTE study. Non-response (n=26/102, 25%) and loss of response over time (n=36/102, 35%) were the main reasons for discontinuation with majority coming from the late responders group (n=25/36, 69%), followed by intolerance (n=19/102, 19%). Seven patients (n=7/102, 7%) discontinued canakinumab as per the physicians’/family decision no longer requiring canakinumab.
Canakinumab tapering/discontinuation
Forty-four (25%) patients received at least three consecutive reduced doses of canakinumab of 2 mg/kg. Of these, 26 (59%) remained on a sustained reduced dose until study end with a median time of follow-up of 25 months. Of these 26 patients, five discontinued canakinumab (no longer requiring study drug) and two discontinued the study (consent withdrawal, n=1; lymphadenitis, n=1). Conversely, of the remaining 18 patients who flared after tapering of canakinumab, 15 regained sJIA control on uptitration of canakinumab to 4 mg/kg/month while three patients did not. In two of those patients disease flare was associated with MAS.
sJIA disease control and response to canakinumab
F igure 2 shows the change in JADAS score over time in the ITT population for the two major subgroups (early and late responders) who entered the LTE from part I and part II of trial 2.

JADAS scores over time in the ITT population for the two major subgroups (early and late responders) who entered the LTE from part I and part II of trial 2 (observed data). n denotes the number of patients available at that time point; horizontal lines represent the cut-offs for JADAS HDA>10.5, LDA≤3.8 and ID≤1. Denominator is equal to 177 patients per the ITT principle. The upper and lower error bars represent the third (q3) and first (q1) quartiles, respectively. Patients who entered the LTE are divided into two subgroups: (1) early responders, defined as patients who had successfully completed the glucocorticoid tapering in part I of trial 2 as per protocol and who were randomised to the withdrawal part; (2) late responders, defined as patients who moved directly from the open-label part of trial 2 and who failed to taper glucocorticoids in part I. All patients who belonged to the full analysis set in trial 2 were taken into account. Only the last available assessment within the given interval was taken into account. Only patients with a value at both BSL and the respective post-BSL time point were included. BSL, baseline; CRP, C-reactive protein; HDA, high disease activity; ID, inactive disease; ITT, intent-to-treat; JADAS, Juvenile Arthritis Disease Activity Score; LDA, low disease activity.
For the patients who entered the LTE from the open-label part of trial 2 (n=48), a decrease in disease activity by a median of 64.6% (n=48) (median change: −24.9 (Q1 −32.7; Q3 −14.9)) and 93.5% (n=25) (median change: −31.8 (Q1 −40.3; Q3 −24.8)) was observed within the 6 months and 2 years of canakinumab treatment, respectively. However, despite improvement on canakinumab, this group of patients continued to remain in JADAS HDA status (JADAS >10.5).
Early responders (n=96) achieved a greater decrease in the JADAS during the study as compared with late responders (mixed model; p<0.01). Notably, in early responders, JADAS improvement by a median of 96.4% (n=95) (median change: −25.1 (Q1 −33.1; Q3 −20.2)) occurred within the first 6 months of canakinumab initiation, and was maintained over at least 2 years (median decrease of 99.2%, n=87) (median change: −27.6 (Q1 −34.3; Q3 −20.6) at 2 years). Overall, from the entire cohort, a total of 79/177 (44.6%) achieved JADAS-LDA/inactive disease (ID) over the first 6 months of canakinumab exposure which increased to 48.6% at 2 years and was sustained until the end of the study.
As shown in figure 3, aJIA-ACR 50/70/90 response rates in the ITT population at 6 months since initiation of canakinumab were 73.4%, 65.5% and 52.0%, respectively. This level of response was maintained up to 3 years with 54.8%, 53.7% and 49.7% of the patients achieving aJIA-ACR 50/70/90 response rates, respectively. ClDACR since initiation of canakinumab was achieved by 32.8% of patients at 6 months, which increased up to 39.5% at 2 years and maintained through (36.7%) at year 3 and progressively decreased to 12.4% in 5 years. The CIDJADAS criteria use yielded similar results.
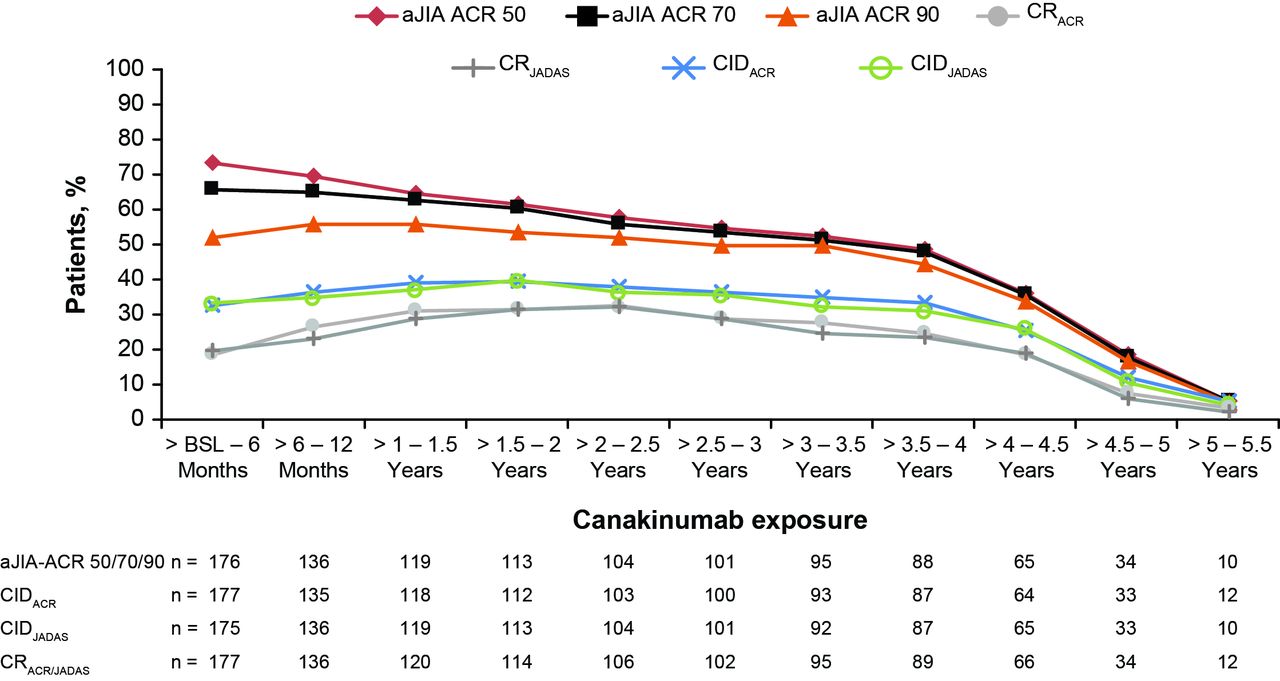
{kind=link}
{kind=link}
{kind=link}
aJIA-ACR responses, CIDACR/CIDJADAS rates and clinical remission on medication rates as per CRACR or CRJADAS over time (observed data). n denotes the number of patients available at that time point; denominator was equal to 177 patients as per the ITT principle in the pivotal study. Clinical remission on medication, defined as CID (ACR or JADAS) for 6 continuous months. aJIA-ACR, adapted juvenile idiopathic arthritis by American (online supplementary appendix) College of Rheumatology criteria; BSL, baseline; CIDACR, clinically inactive disease by ACR criteria; CIDJADAS, clinically inactive disease by Juvenile Arthritis Disease Activity Score criteria; CR, clinical remission; CRACR, CR by American College of Rheumatology criteria; CRJADAS, CR by Juvenile Arthritis Disease Activity Score criteria; ITT, intent-to-treat.
As per figure 3, CRACR criteria were achieved by 18.6% (33/177) of patients within the first 6 months of canakinumab therapy. In these 33 early responding patients, CRACR was reached at a median time of 29 days. The number of patients reaching CRACR further increased to 31.6% after 24 months. During the first 2 years, 71 patients had achieved CRACR on canakinumab with the median time to CR being 213 days. During the study, 80 patients had achieved CRACR (median time=254 days). Consistently similar rates were observed based on CRJADAS (rates are shown in figure 3).
Efficacy analyses were also performed by means of LOCF. (online supplementary figures S1 and S2) show the corresponding LOCF data confirming a trend towards higher level of response over time.
Differences between the ITT and LOCF analyses
Both ITT and LOCF analyses were conducted for the entire study duration of 5 years and showed about 30%–40% achieving ID at year 3, the overall planned study duration. Because the study lasted longer in some countries (until year 5), patients who completed the study as planned at year 3 were counted as discontinuations in the ITT analysis. This resulted in the dissociation observed in the remission rates from year 3 to year 5 between the two methods. While maintenance of response up to year 5 has been demonstrated using LOCF, remission percentages started fading from year 3 onwards in the ITT analysis.
Efficacy analysis of subgroups
A more pronounced decrease in disease activity as measured by the JADAS scores was observed in biologic naïve patients compared with biologic exposed patients at 6 months (92.7% (n=61); median change: −23.0 (Q1 −31.3; Q3 −14.3) vs 75.6% (n=113); median change: −23.4 (Q1 −31.8; Q3 −16.4)) (online supplementary figure 3). Similar trends were observed for the aJIA-ACR response criteria and CIDACR (online supplementary figure 4).
Supplemental material
Supplemental material
Patients treated with canakinumab while on MTX background therapy and those treated with canakinumab alone showed similar JADAS and aJIA-ACR response rates and CIDACR (online supplementary figures S5 and S6).
Glucocorticoid tapering
Among 128/177 (72.3%) patients who were on glucocorticoids at the beginning of trial 2 (median dose of 0.27 mg/kg/day; Q1–Q3: 0.16–0.53 mg/kg/day), 38/128 (29.7%), 51/128 (39.8%) and 20/128 (15.6%) discontinued glucocorticoid therapy by 6 months, 2 years and 5 years of canakinumab therapy, respectively. At the end of the study, 72 patients (56%) remained on glucocorticoid treatment at a median dose of 0.25 mg/kg/day (Q1–Q3: 0.16–0.46 mg/kg/day), with 28 patients (22%) having the dose below 0.2 mg/kg/day level.
As shown in table 1, patients who entered LTE from the double-blind withdrawal part were on lower dose of glucocorticoids (0.17 mg/kg/day) compared with patients who moved from the open-label part I of trial 2 (median dose of 0.36 mg/kg/day). Of the 38 patients who failed to discontinue glucocorticoids at part I of trial 2, ten became steroid free, with another nine patients reaching the below 0.2 mg/kg/day level.
Safety
Overall, the median duration of exposure (canakinumab and placebo) in the study was 3.5 years (Q1, 0.6; Q3, 4.4) corresponding to 476.530 patient-years. The exposure-adjusted incidence rate of AEs was 796.69/100 patient-years (table 2).
Incidence rates of adverse events, by system organ class
The incidence rate of SAEs was 40.68/100 patient-years. Most common SAEs included sJIA flare (5.24/100 patient-years), MAS events (histiocytosis haematophagic in MedDRA terms) and fever either due to infection or sJIA flare (table 3).
Incidence rates of serious adverse events by preferred term (≥2 events)
All MAS cases were adjudicated by an independent adjudication committee for a total of 13 events (2.726/100 patient-years) in 12 patients leading to nine patients discontinuing the study. Three of these were reported earlier5 and the other 10 MAS cases were reported as SAEs in the LTE study. One of the reported MAS events was complicated by pulmonary hypertension and interstitial pneumonia, resulting in patient death in the pivotal study as previously reported,5 and another event was complicated by transfusion-related acute lung injury; acute interstitial pneumonitis, following blood transfusion products for MAS in the LTE study. The second event resolved and the patient discontinued the study. No malignancies, anaphylaxis or anaphylactoid reactions were reported. There were no additional deaths besides the three previously reported. 5
Infections
The incidence of serious infections was 10.28/100 patient-years of exposure and all resolved without sequelae. Most common serious infections were gastroenteritis (1.05/100 patient-years), pneumonia (0.84/100 patient-years), and varicella, subcutaneous abscess, gastrointestinal viral infection, septic shock and streptococcal tonsillitis (0.42/100 patient-years each).
There were four opportunistic infections (toxoplasmosis, cytomegalovirus infection, Salmonella gastroenteritis and adenovirus infection) in one patient, each was adjudicated as such by the adjudication committee; two of these events were suspected to be related to canakinumab by the investigator. All events were moderate in severity and resolved following treatment. No tuberculosis cases were reported.
Laboratory abnormalities
Transient neutropenia was reported in 18 patients (grade 3 (n=17); grade 4 (n=1)). Eleven neutropenia events occurred in patients on MTX background and three were associated with mild infections (pharyngitis, molluscum contagiosum, nasopharyngitis and otitis media), respectively. None of the patients discontinued canakinumab due to neutropenia but two patients discontinued MTX.
Besides laboratory testing that reflect the anti-inflammatory effects of canakinumab, other clinical chemistry parameters remained largely unchanged. CRP and fibrinogen decreased markedly by week 2 of canakinumab therapy and low levels were maintained over time.
Immunogenicity
Post-treatment ADAs were detected in five patients. ADAs were all non-neutralising and their presence had no effect on PK of canakinumab; the observed trough canakinumab concentrations in presence of ADAs were comparable to those without ADAs. All but one of the five patients were on background treatment with MTX.
Discussion
Here, we report the long-term efficacy and safety data of canakinumab in patients with sJIA with active systemic features and arthritis enrolled in the pivotal phase III trials.5 There was a marked, rapid improvement of sJIA activity at 6 months, which was maintained for up to 5 years and allowed for the marked reduction or even discontinuation of glucocorticoids in the majority of patients.
Due to the long duration of the study, canakinumab efficacy seems best described by the level of disease control as measured by the JADAS. Exploratory analysis suggested that early response to canakinumab leads to a better long-term favourable outcome. As such, patients who entered the LTE study from the double-blind, placebo-controlled part because they responded to canakinumab quickly and successfully tapered glucocorticoids, fared better than late responders, that is, patients who moved directly from part I of trial 2 because they failed to respond to canakinumab initially or were unable to taper glucocorticoids as per protocol. These data were further corroborated by the CR data, which showed a trend towards better improvement in the subgroup of patients who were naïve to biologics.
Glucocorticoid discontinuation was possible for 44% of the patients, some rapidly and some in the long term, suggesting a continuous glucocorticoid tapering effect of canakinumab.
The limited therapeutic benefits of MTX in sJIA were confirmed by similar response rates to canakinumab irrespective of MTX background therapy. Thus, canakinumab/MTX combination therapy is unlikely expected to improve sJIA control versus using canakinumab alone.
Most children who reached CID/LDA level after canakinumab initiation who were randomised to placebo and flared in the double-blinded part of trial 2 regained the LDA status on retreatment with canakinumab. This observation may suggest that canakinumab therapeutic benefits can be recaptured after withdrawal or interruption of medication in children who previously responded well to canakinumab. Based on the time to flare in the double-blinded part of the study, a CID/LDA state would be expected to be maintained longer than the half-life of the drug. Further, these data also support that the withdrawal study design with an event-driven approach as was chosen for trial 2 did not expose children switched to placebo to worse long-term outcomes than children who were randomised to continue canakinumab.
The long-term use of canakinumab was well tolerated and consistent with the safety profile that has been reported in other canakinumab trials.5 Infections were the most common AEs. Despite disease control, new MAS events occurred while on canakinumab therapy. This is consistent with the previous reports,33 which suggested that canakinumab treatment does not seem to alter the risk of MAS in patients with sJIA, regardless of the response of sJIA to canakinumab. Of note, the MAS adjudication committee was established before the EULAR/ACR/PRINTO MAS classification criteria were published.33
A limitation of this study may be the overall low retention rate with 58% of the patients discontinuing canakinumab over the 5 years, which is typically expected in a study with such long follow-up. However, LOCF was carried out in addition to the observed data to address this shortcoming and possible artificial increasing percentages of response rates over time. The higher discontinuation rate in the late responders group as opposed to the early responders group further supports the notion of early response being used as a predictor factor of long-term outcome by the physicians when considering canakinumab change to another treatment. In addition, this LTE study included patients with a wide range of canakinumab treatment durations due to the adaptive design chosen for trial 2 to limit placebo and corticosteroid exposures to patients. To address the limitations, another cohort of sJIA is being studied, where canakinumab was started in patients with and without fever at baseline (analysis in progress). Further, a dedicated dose reduction/dose interval prolongation study in patients with sJIA who are canakinumab responders is ongoing (NCT02296424).
In conclusion, response to canakinumab treatment was sustained or improved up to 5 years in patients with sJIA with active systemic features and arthritis and was associated with glucocorticoid discontinuation. Early response seemed to be a predictive factor of long-term outcome enabling physicians to incorporate in their decision-making the time to response in the consideration of changing canakinumab to another treatment. No new safety findings were observed on long-term use of canakinumab.
Supplemental material
Supplemental material
Supplemental material
Supplemental material
Acknowledgments
The authors thank all additional investigators for their participation in the study: Wolfgang Emminger, Wien, Austria; Laurence Goffin, Laeken, Belgium; Rik Joos, Gent, Belgium; Bernard Lauwerys, Bruxelles, Belgium; Carine Wouters, Leuven, Belgium; Claudio Len, Sao Paulo, Brazil; Maria Hilário, São Paulo, Brazil; Sheila Oliveira, Rio de Janeiro, Brazil; Flavio Sztajnbok, Rio de Janeiro, Brazil; Elie Haddad, Montreal, QC, Canada; Lori Tucker, Vancouver, Canada; Kristin Houghton, Vancouver, BC, Canada; Brigitte Bader Meunier, Paris, France; Richard Mouy, Paris, France; Isabelle Kone-Paut, Le Kremlin-Bicêtre, Bicêtre, France; Angelika Thon, Hannover, Germany; Elisabeth Weibarth-Riedel, Hamburg, Germany; Hartwig Lehmann, Gießen, Germany; Ivan Foeldvari, Hamburg, Germany; Jasmin Kummerle-Deschner, Tübingen, Germany; Johannes-Peter Haas, Germany; Michael Frosch, Muenster, Germany; Nikolay Tzaribachev, Germany; Thomas Lutz, Heidelberg, Germany; Tilmann Kallinich, Berlin, Germany; Giorgios Chrousos, Athens, Greece; Maria Trachana, Thessaloniki, Greece; Olga Vougiouka, Ampelokipi, Greece; Riva Brik, Haifa, Israel; Liora Harel, Petach-Tikvah, Israel; Shay Padeh, Ramat Gan, Israel; Yosef Uziel, Kfar-Sava, Israel; Yackov Berkun, Ramat Gan, Israel; Maria Alessio, Napoli, Italy; Rolando Cimaz, Firenze, Italy; Valeria Gerloni, Milano, Italy; Berit Flato, Oslo, Norway; Manuel Ferrandiz, Breña, Peru; Lidia Rutkowska-Sak, Warszawa, Poland; Ekaterina Alekseeva, Moscow, Russia; Evgeny Nasonov, Moscow, Russia; Vyacheslav Chasnyk, Saint-Petersburg, Russia; Inmaculada Calvo, Valencia, Spain; Maria Luz Gamir, Madrid, Spain; Juan Carlos Lopez Robledillo, Madrid, Spain; Bo Magnusson, Stockholm, Sweden; Michael Hofer, Lausanne, Switzerland; Huri Ozdogan, Istanbul, Turkey; Muferet Erguven, Istanbul, Turkey; Seza Ozen, Ankara, Turkey; Alice Chieng, Manchester, UK; Liza McCann, Liverpool, UK; Clarissa Pilkington, London, UK; Nicholas Wilkinson, Oxford, UK; Patricia Woo, London, UK; Taunton Southwood, Birmingham, UK; Gloria Higgins, Columbus, OH, USA; Daniel Kingsbury, Portland, OR, USA; Jorge Lopez-Benitez, Boston, MA, USA; Katherine Marzan, Los Angeles, CA, USA; Paula Morris, Little Rock, AR, USA; Kenneth Schikler, Louisville, KY, USA. Medical writing and editorial assistance was provided by Divya Chandrasekhar, PhD, of Novartis Healthcare, Hyderabad, India, which was funded by Novartis Pharma, Basel, Switzerland.
References
Footnotes
NR and HIB contributed equally.
Handling editor Josef S Smolen
Contributors The study was designed jointly by academic authors (NR, HIB, AM and DJL) and Novartis, with data collected by PRINTO/PRCSG investigators. The first and subsequent versions of the manuscript were written by NR and HIB, edited by AM and DJL and revised critically by all remaining coauthors. All authors attest to the completeness and veracity of data and data analyses. Consistency in reporting the study data to healthcare authorities and institutional review boards was ensured by Novartis. All authors had full access to study data, reviewed and revised the manuscript and approved the final version to be published. All authors were involved in the decision to submit the manuscript for publication and had the right to accept or reject comments or suggestions.
Funding This study was funded by Novartis Pharma.
Competing interests NR: consultant and speaker’s bureaus from AbbVie, Ablynx, Amgen, AstraZeneca, Baxalta Biosimilars, Biogen Idec, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Eli-Lilly, EMD Serono, Gilead Sciences, Janssen, MedImmune, Novartis, Pfizer, R-Pharm, Roche, Sanofi, Servier, Takeda; NR works as a full-time public employee of the public hospital Istituto Giannina Gaslini, which has received contributions from Bristol-Myers Squibb, Hoffman-La Roche, Janssen, Novartis, Pfizer, Sobi for the coordination activity of the PRINTO network. HIB: consultant: AstraZeneca, Bristol-Myers Squibb, Genentech, Janssen, Novartis, Pfizer, Sanofi, Takeda; speaker’s bureaus: Genentech, Novartis Pierre Quartier; has received consultant fees from Novimmune, Novartis and SOBI; has received speaker’s bureaus from Abbvie, BMS, Chugai-Roche, Lilly, Novartis, Pfizer and Sobi; has acted as coordinator or investigator in clinical trials for Abbvie, BMS, Chugai-Roche, Novartis, Sanofi, Sobi; has acted as member of a data monitoring board committee for Sanofi. NMW has received grant/research support from AbbVie; has received consultant fees from AbbVie, Sobi and Novartis. GH has received speaker’s bureaus from AbbVie, Boehringer Ingelheim, Chugai, MSD, Novartis, Pfizer, Roche and Sobi; has received scientific grants from AbbVie, Chugai, MSD, Novartis, Pfizer and Roche. OK has received speaker’s bureaus from Novartis, Roche, Pfizer and Abbvie. RS has received consultant fees from Novartis, Sobi and Novimmune. JLA has received consultant fees and speaker’s bureaus from AbbVie, Gebro, Novartis, Pfizer, Roche, Sanofi and Sobi. RC has received consultant fees and speaker’s bureaus from Bristol-Myers Squibb, Janssen, Novartis, Pfizer, Sanofi, Roche, Lilly and GlaxoSmithKline. HEF has acted as member of advisory boards for AbbVie, Novartis, Pfizer, Sanofi and Sobi; has received unrestricted educational bursaries from Pfizer, Genzyme, BioMarin and Sobi to develop educational resources for healthcare professionals. DF has received grant/research support from Pfizer and Novartis; has received consultant fees from Novartis, Pfizer, Chugai-Roche and Sobi; has received speaker’s bureaus from Novartis. SCR has received grants for clinical research from Novartis. AVR has received honoraria and participated in Advisory Boards for Novartis. RT has received lecture fees from Pfizer and Bristol-Myers Squibb. JL received fees from Novartis for conducting the statistical analysis. EV is an employee of Novartis Pharma, Basel, Switzerland. AM has no conflicts of interest to declare since March 2016 when he became the Scientific Director of the Istituto Giannina Gaslini, because this role does not allow him to render private consultancy resulting in personal income; consultant on behalf of the Istituto Giannina Gaslini: AbbVie, Boehringer, Novartis, R-Pharm. Istituto Giannina Gaslini has received contributions from Bristol-Myers Squibb, Hoffman-La Roche, Janssen, Novartis, Pfizer, Sobi for the coordination activity of the PRINTO network. DJL has received grant/research support from AbbVie, Bristol-Myers Squibb, NIH, Pfizer, Roche; speaker’s bureaus from Genentech; consultant of AbbVie, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Genentech, GlaxoSmithKline, Janssen, Johnson & Johnson, Novartis, Takeda, UCB. EU, FC, JB, MF, MF-d, RB and TC have nothing to disclose.
Patient consent Parental/guardian consent obtained.
Ethics approval The study was conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonisation Guidelines for Good Clinical Practice and local regulations. An institutional review board or independent ethics committee at each site approved the protocol, consent form and any other written information provided to patients or their legal representatives.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All data generated or analysed are included in this article and the supplementary information files.