Article Text

Abstract
Objectives Methotrexate (MTX) is the anchor drug for treatment of rheumatoid arthritis (RA), but the mechanism of its anti-inflammatory action is not fully understood. In RA, macrophages display a proinflammatory polarisation profile that resembles granulocyte-macrophage colony-stimulating factor (GM-CSF)-differentiated macrophages and the response to MTX is only observed in thymidylate synthase+ GM-CSF-dependent macrophages. To determine the molecular basis for the MTX anti-inflammatory action, we explored toll-like receptor (TLR), RA synovial fluid (RASF) and tumour necrosis factor receptor (TNFR)-initiated signalling in MTX-exposed GM-CSF-primed macrophages.
Methods Intracellular responses to TLR ligands, TNFα or RASF stimulation in long-term low-dose MTX-exposed human macrophages were determined through quantitative real-time PCR, western blot, ELISA and siRNA-mediated knockdown approaches. The role of MTX in vivo was assessed in patients with arthritis under MTX monotherapy and in a murine sepsis model.
Results MTX conditioned macrophages towards a tolerant state, diminishing interleukin (IL)-6 and IL-1β production in LPS, LTA, TNFα or RASF-challenged macrophages. MTX attenuated LPS-induced MAPK and NF-κB activation, and toll/IL-1R domain-containing adaptor inducing IFN-beta (TRIF1)-dependent signalling. Conversely, MTX increased the expression of the NF-κB suppressor A20 (TNFAIP3), itself a RA-susceptibility gene. Mechanistically, MTX-induced macrophage tolerance was dependent on A20, as siRNA-mediated knockdown of A20 reversed the MTX-induced reduction of IL-6 expression. In vivo, TNFAIP3 expression was significantly higher in peripheral blood cells of MTX-responsive individuals from a cohort of patients with arthritis under MTX monotherapy, whereas MTX-treated mice exhibited reduced inflammatory responses to LPS.
Conclusions MTX impairs macrophage proinflammatory responses through upregulation of A20 expression. The A20-mediated MTX-induced innate tolerance might limit inflammation in the RA synovial context, and positions A20 as a potential MTX-response biomarker.
- methotrexate
- rheumatoid arthritis
- dmards (synthetic)
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic, systemic inflammatory disorder that primarily affects synovial joints. Predominant cytokines in RA pathogenesis are TNFα, interleukin (IL)-6 and IL-1β, which are mainly produced by macrophages and act locally and systemically.1–3 In fact, macrophages accumulate in the synovium of RA joints, where they exhibit destructive and remodelling potential and contribute considerably to inflammation and joint destruction.4 Importantly, reduction in the number of macrophages in the synovium constitutes a biomarker for response to treatment in patients with RA.5
GM-CSF induces macrophage differentiation from haematopoietic progenitor cells and is a key driver of tissue inflammation.6 GM-CSF was one of the first cytokines detected in human synovial fluid from inflamed joints and several lines of data suggest that GM-CSF strongly influences the development and pathogenesis of RA.6 GM-CSF-deficient mice are protected from developing collagen-induced arthritis and blockade of GM-CSF reduces the severity of established disease in wild-type mice, thus supporting a key role for GM-CSF in the initiation and development of inflammatory arthritis. Moreover, overexpression or injection of GM-CSF is associated with flares of arthritis and patients receiving GM-CSF after chemotherapy showed exacerbation of established RA.7 In line with all the above findings, macrophages from patients with active RA display a transcriptomic and phenotypical proinflammatory polarisation profile that resembles GM-CSF-differentiated macrophages.8 According to the role of GM-CSF in RA, phase I and II clinical trials targeting GM-CSF or GM-CSF receptor-α in RA have shown rapid and sustained clinical responses without major safety concerns.9–11
The folic acid antagonist methotrexate (MTX) is the disease modifying anti-rheumatic drug of first choice in the treatment of early and established RA, and shows good efficacy, with 40% of patients with RA achieving an American College of Rheumatology score 50 (ACR50) response.12 13 MTX blocks different enzymes of the folate or one-carbon metabolism. While MTX blocks dihydrofolate reductase, polyglutamated MTX potently inhibits thymidylate synthase (TS), and both enzymes are crucial for the de novo biosynthesis of purines and pyrimidines required for DNA replication and cellular proliferation.13 In patients with RA, MTX treatment is associated with a significant decrease in serum IL-6 and IL-8.14 However, despite the beneficial effect of MTX in current RA therapies, its mechanism of action as an anti-inflammatory drug remains inconclusive. Different cellular-specific mechanisms have been proposed to explain the inhibition of NF-κB, the central regulator of proinflammatory cytokine gene expression. Previous studies on T lymphocytes and fibroblast-like synoviocytes have found that MTX inhibits NF-κB activity via tetrahydrobiopterin (BH4) depletion, lincRNA-p21 increase or adenosine receptor activation, respectively.15 16 Although MTX restores NF-κB activity in peripheral blood mononuclear cells (PBMCs) from patients with RA,15 no information is available on the effect of clinical doses of MTX on NF-κB in human macrophages, whose transcriptome is significantly modulated after low-dose MTX exposure.17
We have also previously described that MTX exclusively targets proinflammatory TS+ GM-CSF-primed macrophages.17 In an attempt to determine the molecular mechanism underlying the anti-inflammatory actions of MTX, we have explored the inflammatory response of MTX-exposed GM-CSF-primed macrophages. We now report that long-term low-dose MTX treatment modifies macrophage response to TLR ligands or TNFα stimulation by increasing TNFAIP3 (A20) expression in macrophages, and that MTX conditions macrophages for impaired responses towards pathogen agents like LPS, TNFα or RA synovial fluid (RASF).
Methods
Detailed methods are supplied in the online supplementary file.
Supplementary file 1
Human PBMCs were isolated from buffy coats from normal donors. Monocytes were purified from PBMCs by magnetic cell sorting using CD14 microbeads (Miltenyi) and were cultured at 0.5×106 cells/mL for 7 days in Roswell Park Memorial Institute (RPMI) supplemented with 10% fetal calf serum, and containing GM-CSF (1000 U/mL) to generate GM-CSF-polarised macrophages (GM-MØ). Independent preparations of monocytes were unexposed or exposed once to MTX (50 nM)18 19 and differentiated to GM-MØ for 7 days. MTX is a drug given weekly to patients with RA and we followed in vitro the patient schedule.12 LPS (10 ng/mL, 01111:B4 strain), LTA (5 µg/mL), TNFα (20 ng/mL) and RASF were added for the indicated time points to 7-day fully differentiated GM-MØ and were analysed by quantitative real-time RT-PCR, immunobloting or ELISA. RASF were obtained from patients with active knee arthritis, confirmed by a highly cellular synovial fluid, and were heterogeneous regarding demographic, disease characteristics and previous RA therapy. For in vivo MTX-cross tolerance, C57BL/6J mice between 6 and 8 weeks of age (n=6 mice/group) received phosphate-buffered saline (PBS) or MTX intraperitoneally (2 mg/kg). Seven days later, intraperitoneal LPS (9 mg/kg) was administered and serum collected after 4 hours for IL-6 and TNFα determination by ELISA. Patients with early arthritis in MTX monotherapy were recruited from the Princesa Early Arthritis Register Longitudinal study. Informed consent is received from the patient.20 Statistical analysis was performed using paired Student’s t-test and a P value <0.05 was considered significant (*P<0.05, **P<0.01, ***P<0.001).
Results
Long-term low-dose MTX treatment diminishes LPS, LTA, TNFα and RASF-induced proinflammatory cytokine production in human macrophages
A variety of TLR ligands, either endogenous or of microbial origin, are present within RA synovia.21 To determine the role of MTX on macrophage TLR4 activation, monocytes were exposed to a single dose of MTX before initiation of the GM-CSF-driven differentiation process, and later challenged with LPS at the 7-day culture (figure 1A). MTX pretreatment diminished LPS-induced IL-6 expression at the mRNA and protein levels (figure 1B). Similarly, MTX pretreatment reduced LPS-induced TNFα, IL-12p40 and IL-1β production although with different kinetics (figure 1C and online supplementary figure S1). By contrast, the expression of other LPS-responsive macrophage genes22 was either unaltered (IFIT2, PKIG) or increased (CCR7) on MTX pretreatment (online supplementary figure S1), thus indicating that proinflammatory cytokine production is selectively inhibited in MTX-exposed macrophages. The specificity of MTX to attenuate LPS-dependent proinflammatory cytokine production in macrophages was analysed in the presence of folinic acid (FA). FA restored LPS-induced IL-6 secretion in MTX-treated macrophages, indicating that MTX-induced effects are mediated through blocking the one-carbon metabolism (figure 1D). Similar to TLR4 activation, MTX-treated macrophages exhibited a lower level of LTA-dependent IL-6 and IL-1β gene expression and protein secretion (figure 1E). More importantly, analogous findings were observed when macrophages were challenged with TNFα, the predominant inflammatory cytokine found in RA joints1: MTX-treated macrophages exhibited a lower expression of TNFα-induced IL6 and IL1B mRNA and IL-6 and IL-12p40 secretion than untreated macrophages (figure 1F), thus indicating that MTX attenuates proinflammatory cytokine expression in response to TLR4 and TLR2 ligands as well as after TNFα stimulation.

MTX alters TLR4, TLR2 and TNFα responsiveness in GM-CSF-primed macrophages. (A) Schematic representation of the experiments. Monocytes were exposed to 50 nM MTX at the beginning of the 7-day macrophage differentiation process with GM-CSF and challenged with LPS (10 ng/mL), LTA (5 µg/mL) or TNFα (20 ng/mL) on day 7. Cells (GM-MØ) were assayed at time points poststimulation. (B) Expression of IL-6 by qRT-PCR (left) or ELISA (right) by monocytes differentiated with GM-CSF in the absence or presence of MTX and challenged with LPS for 48 hours. Mean±SEM of four independent donors are shown (*P<0.05, **P<0.01). (C) Production of TNFα or IL-12p40 by monocytes differentiated with GM-CSF in the absence or presence of MTX and challenged with LPS for 48 hours. Mean±SEM of four independent donors are shown (*P<0.05, **P<0.01). (D) Production of IL-6 by monocytes differentiated with GM-CSF in the absence or presence of MTX or FA (1 µM) and challenged with LPS for the indicated time points, as determined by ELISA. Mean±SEM of three independent donors are shown (*P<0.05, **P<0.01). (E–F) Expression of IL-6, IL-1β and IL-12p40 by qRT-PCR (upper panels) or ELISA (lower panels) by monocytes differentiated with GM-CSF in the absence or presence of MTX and challenged with LTA (E) or TNFα (F) for the indicated time points. Mean±SEM of three (E) and five (F) independent donors are shown (*P<0.05). FA, folinic acid; IL, interleukin; Mo, monocytes; MTX, methotrexate; qRT-PCR, quantitative real-time PCR.
Finally, to determine whether MTX-exposed macrophages are also refractory to the effect of the RA synovial environment, MTX-treated macrophages were exposed to RASF from patients with active disease. RASF-induced IL6 mRNA was attenuated in MTX-treated macrophages although the effect of MTX differed among tested RASF (figure 2). Altogether, these results indicate that MTX diminishes the production of RA-associated cytokines in human macrophages exposed to stimuli known to be present in RA joints (TLR2 and TLR4 ligands, TNFα and RASF).
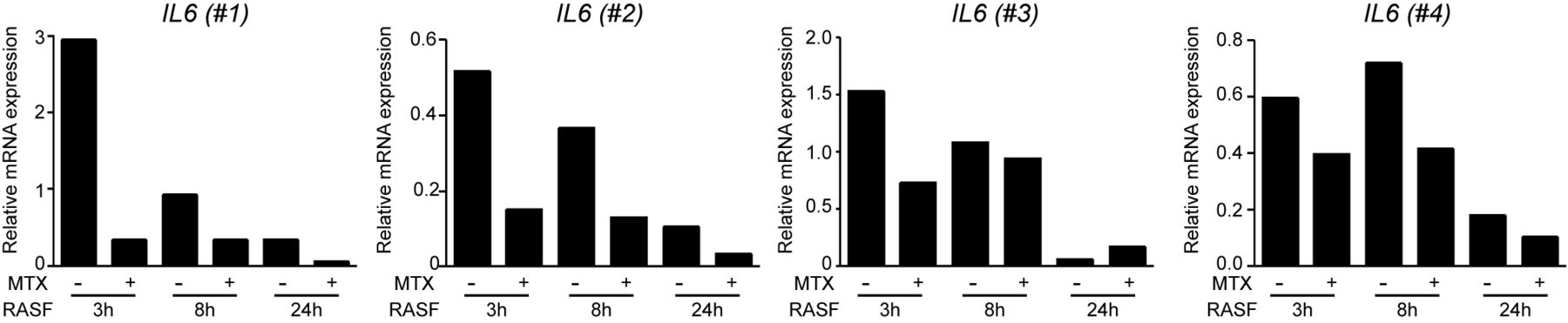
MTX diminishes RASF IL6 induction in GM-CSF-primed macrophages. Expression of IL6 by quantitative real-time PCR by monocytes differentiated with GM-CSF in the absence or presence of MTX and challenged with 25% synovial fluid from patients with active RA (RASF) on day 7 for the indicated time points (*P<0.05). Independent macrophage donors (nos 1–4) were challenged with RASF from two different patients with RA (donor no. 1 with RASF-A and donors no. 2, no. 3 and no. 4 with RASF-B). IL, interleukin; MTX, methotrexate; RA, rheumatoid arthritis; RASF, RA synovial fluid.
TNFAIP3 is an MTX response gene and is involved in MTX-induced tolerance in macrophages
The above results suggested that low-dose MTX renders macrophages less responsive to a subsequent stimulation by proinflammatory stimuli and that MTX might impose a state of desensitisation in macrophages that resembles the ‘LPS tolerance’ phenomenon.23 Interestingly, and in line with the potential acquisition of a tolerance state in MTX-treated macrophages, Gene Set Enrichment Analysis (GSEA) on the transcriptome of MTX-treated macrophages (GSE71253) revealed that long-term MTX treatment promotes a significant upregulation of the ‘TNFα signalling via NF-κB’ (false discovery rate (FDR) q value=0.0001) and ‘inflammatory response’ (FDR q value=0.0001) gene sets (figure 3A).17 24 In agreement with the GSEA prediction, MTX-treated monocytes led to the generation of macrophages with a significantly higher IL1B, IL1A and IL6 mRNA expression (figure 3B).

MTX suppresses TLR4 and TNFR signalling and induces TNFAIP3 (A20) in macrophages. (A) Gene Set Enrichment Analysis results for MTX-treated versus untreated macrophages for 7 days indicating the normalised enrichment score and the false discovery rate. (B) Relative expression of IL1B, IL1A, IL6 and TNFA in MTX-treated (Mo+GM(MTX)) and untreated (Mo+GM) GM-CSF-primed macrophages (*P<0.05, adjusted P value), as determined by microarray (left) and qRT-PCR (right). (C) Immunoblot analysis of pp38, pJNK and pERK, p38, JNK, extracellular signal-regulated kinase (ERK) and IκBα by monocytes differentiated with GM-CSF in the absence or presence of MTX for 7 days and challenged with LPS (left) or TNFα (right) for the indicated time points. A representative experiment of four independent donors is shown. (D) Basal and LPS-induced NF-κB-dependent transcriptional activity in MTX-treated and untreated GM-MØ. Mean±SEM of the relative NF-κB luciferase activity (compared with Renilla luciferase activity) of five independent experiments are shown (*P<0.05). (E) Immunoblot analysis of pStat1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (left), and CXCL10 secretion (right) by monocytes differentiated with GM-CSF in the absence or presence of MTX for 7 days and challenged with LPS for 2 hours (left) or 24 hours (right). (F) Relative expression of the genes encoding molecules involved in tolerance (black, upregulated in LPS tolerance; white, downregulated in LPS tolerance) in MTX-treated (Mo+GM(MTX)) and untreated (Mo+GM) GM-CSF-primed macrophages (*P<0.05, adjusted P value). (G) Expression of TNFAIP3 by monocytes differentiated with GM-CSF in the absence or presence of MTX, as determined by qRT-PCR. Mean±SEM of three independent donors are shown (*P<0.05, ***P<0.001). Immunoblot analysis of A20 and GAPDH in monocytes differentiated with GM-CSF in the presence of MTX at the indicated time points. (H) Monocytes differentiated with GM-CSF in the absence or presence of MTX for 5 days were transfected with control siRNA (siC) or siRNA for A20 (siA20) and 48 hours later, cells were then stimulated with LPS for 3 hours, and expression levels of A20, GAPDH (immunoblot) and IL-6 (ELISA) were detected. IL, interleukin; Mo, monocytes; MTX, methotrexate; qRT-PCR, quantitative real-time PCR.
To address whether MTX promotes ‘bona fide’ innate tolerance in macrophages, and since tolerant cells exhibit a lower level of MAPK and NF-κB activation on TLR stimulation,23 24 we first determined the levels of LPS and TNFα-induced MAPK and IκBα activation in MTX-treated macrophages. MTX pretreatment reduced the activation of p38 and jun amino-terminal kinase (JNK) in response to LPS and TNFα (figure 3C). Moreover, a higher level of IκBα was detected in LPS and TNFα-exposed MTX-pretreated macrophages (figure 3C), indicating that the LPS and TNFα-induced activation of both MAPK and NF-κB was impaired in MTX-treated macrophages. These results were further confirmed after analysis of the LPS-induced NF-κB-dependent transcription (figure 3D), as MTX-treated macrophages exhibited lower LPS-induced NF-κB-dependent transcriptional activity than untreated GM-MØ. Therefore, MTX treatment conditions macrophages for an impaired NF-κB transcriptional activity after LPS stimulation. The TRIF-dependent pathway, implicated in mediating the type I interferon activation of the LPS signalling,25 was also explored. Since MTX-treated cells exhibited lower C-X-C motif chemokine ligand 10 (CXCL10) secretion and phosphorylated signal transducer and activator of transcription 1 (p-Stat1) activation in response to LPS than untreated cells (figure 3E), MTX appears to exert its effects also via the TRIF pathway.
Next, we determined the expression of regulators of TLR-induced cytokine production that have been previously implicated in LPS tolerance.23 Long-term MTX treatment significantly increased the expression of TNFAIP3, decreased TLR4 and TLR2 and did not modulate IRAK3 (IRAK-M), INPP5D (SHIP1), SOCS1, SOCS3 and PELI3 mRNA expression in GM-MØ (figure 3F).23 24 26 27 Reduction of TLR2 and TLR4 by MTX was modest at the mRNA level and not observed in all donors at the protein level (online supplementary figure S2). By contrast, TNFAIP3 induction by MTX was observed in all donors examined. TNFAIP3 codes the A20 protein, a ubiquitin-modifying enzyme that acts as a pivotal NF-κB suppressor after TLRs or TNFR stimulation.28 Kinetic studies revealed that TNFAIP3 expression increased 5 days after MTX addition in GM-CSF-primed macrophages (figure 3G), thus suggesting a role for A20 in MTX-induced tolerance. To explore whether this is the case, we determined the effect of silencing TNFAIP3 expression. siRNA-mediated TNFAIP3 knockdown in MTX-treated GM-MØ significantly restored IL-6 secretion in response to LPS, as macrophages with lower A20 expression yielded significantly higher levels of LPS-induced IL-6 (figure 3H). Therefore, MTX-induced A20 contributes to the reduced LPS-induced IL-6 expression seen in MTX-treated macrophages, suggesting its involvement in the MTX-induced tolerance.
MTX-dependent expression of TNFAIP3 involves TS inhibition and TP53 activation
To explore the role of MTX on TNFAIP3 expression, we first determined the sensitivity of the MTX-dependent TNFAIP3 upregulation to FA. TNFAIP3 induction by MTX was inhibited by the simultaneous addition of FA, indicating that MTX-triggered TNFAIP3 induction relies on blocking one-carbon metabolism (figure 4A). However, FA in the clinic is usually prescribed 24 hours after MTX treatment.29 As expected, FA did not reversed MTX-induced TNFAIP3 expression when added 24 hours after MTX, showing that FA does not block the potential beneficial clinical effects of TNFAIP3 induction by MTX in RA (figure 4B).

MTX-dependent expression of TNFAIP3. TNFAIP3 expression by monocytes differentiated with GM-CSF in the absence or presence of MTX (50 nM), FA or both, as determined by qRT-PCR. In (A), FA (1 µM) was added simultaneously with MTX and in (B) FA (50 nM, concentration of FA found in the serum of patients with RA) was added 24 hours after MTX treatment, as indicated. Mean±SEM of three independent donors are shown (*P<0.05). (C) TNFAIP3 mRNA expression on GM-MØ transfected with control siRNA (siC) or siRNA for TYMS (si8, si9) and exposed to MTX for 48 hours, as determined by qRT-PCR. Results are expressed as fold induction with MTX. Mean and SEM of six independent donors are shown (*P<0.05). (D) TNFAIP3 expression on GM-MØ transfected with control siRNA and siRNA for TYMS and exposed to PFT (50 µM) for 48 hours, as determined by qRT-PCR. Results are expressed as fold induction, which indicates the expression of TNFAIP3 in siRNA TYMS-transfected relative to siRNA control cells, and untreated or treated with PFT. Mean and SEM of six independent donors are shown. FA, folinic acid; IL, interleukin; Mo, monocytes; MTX, methotrexate; PFT, pifithrin-α; qRT-PCR, quantitative real-time PCR; RA, rheumatoid arthritis; TYMS, thymidylate synthetase.
Unlike the rapid induction of A20 in response to inflammatory stimuli that takes place 30–60 min after NF-κB activation,28 a slow induction of A20 was observed in MTX-exposed macrophages (figure 3G). We have previously demonstrated that the mechanism involved in MTX response in proinflammatory macrophages relies on TS inhibition and p53 activation, what led us to assess whether the TS/p53 axis is involved in MTX-induced A20 expression.17 Assessment of the underlying mechanism revealed that MTX-triggered TNFAIP3 induction in GM-MØ was significantly diminished (40%) after TS silencing with two different small interfering RNA (figure 4C), thus indicating that MTX-triggered TNFAIP3 upregulation is partly dependent on TS expression. Moreover, knockdown of TS sufficed to increase TNFAIP3 mRNA expression in GM-MØ, an effect that was prevented in the presence of the p53 inhibitor pifithrin-α (figure 4D). Therefore, MTX induces a tolerant state in macrophages by upregulating the expression of TNFAIP3 in a TS/p53-dependent pathway.
MTX-induced tolerance in vivo
To address the relevance of the MTX-induced tolerance in vivo, mice were treated with MTX for 7 days, mimicking the patient schedule therapy currently in use,12 and challenged with an intraperitoneal injection of LPS before determination of the serum concentrations of IL-6 and TNFα (figure 5A). LPS-induced serum IL-6 and TNFα significantly diminished in MTX-pretreated mice (figure 5B, C), whose white blood cell counts did not differ significantly from those of untreated mice (not shown). The same results were obtained when MTX was injected once a week for 4 weeks (not shown). Therefore, and in agreement with its ability to limit macrophage responses to LPS in vitro, MTX is capable of inducing a state of tolerance in mice in vivo.

Effect of low-dose MTX in LPS tolerance in vivo. (A) Schematic representation of the experiment. C57BL/6J mice received an intraperitoneal injection of either saline or MTX (2 mg/kg). Seven days later, intraperitoneal LPS (9 mg/kg) was administered and serum collected after 4 hours for IL-6 (B) and TNFα (C) determination by ELISA. Data represent the results of two independent experiments using a total of 12 mice per group. Mean±SEM of 12 mice per group are shown (*P<0.05, **P<0.01). MTX, methotrexate.
TNFAIP3 mRNA expression increases in early arthritis patients responders to MTX
To evaluate the association between MTX treatment and TNFAIP3 expression in patients with arthritis, we determined TNFAIP3 mRNA levels in PBMCs from patients with early arthritis at baseline and during 1 year of MTX monotherapy (see online supplementary table S1). We found that TNFAIP3 mRNA expression levels tend to increase along the follow-up (figure 6A, model 1 in online supplementary table S2). Furthermore, TNFAIP3 mRNA expression significantly increased only in those patients who respond to MTX therapy (figure 6B,C, model 2 and model 3 in online supplementary table S2). In fact, being or not an MTX responder explained 20% of the variability of TNFAIP3 expression (R2 m=0.196, model 3 in online supplementary table S2). Altogether, these results indicate that TNFAIP3 expression is higher in MTX responder patients with arthritis, suggesting that TNFAIP3 might be an MTX responsiveness biomarker, although these findings require confirmation in larger cohorts.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of MTX on TNFAIP3 mRNA expression of peripheral blood mononuclear cells from patients with early arthritis. (A) TNFAIP3 expression along the follow-up (n=17 patients; online supplementary table S2). (B) TNFAIP3 expression between responder and non-responder patients along the follow-up (n=17 patients). The variable that explained TNFAIP3 expression was being responder to MTX (P<10−4, model 3 in online supplementary table S2). (C) TNFAIP3 expression between responder and non-responder patients untreated or treated with MTX (n=15 visits in non-responder and n=35 visits in responder patients). MTX, methotrexate.
Discussion
Weekly administered MTX is the main starting therapy and the anchor drug for the treatment of RA, cutaneous psoriasis and psoriatic arthritis.30 However, the exact mechanism underlying the anti-inflammatory actions of MTX remains not fully understood.13 We have previously shown that MTX triggers the acquisition of a proinflammatory gene profile in human GM-CSF-primed macrophages.17 We now report that long-term low-dose MTX conditions GM-CSF-primed macrophages towards a tolerance state that renders them less responsive to TLR ligands, TNFα and RASF stimulation. Mechanistically, MTX reduces LPS and TNFα-dependent MAPK activation, IκBα degradation, NF-κB activity and proinflammatory cytokine production in human macrophages. In addition, MTX increases A20 expression, a pivotal NF-κB suppressor whose knockdown impairs the MTX-induced tolerance effect. In line with these findings, MTX-treated mice exhibit a reduced inflammatory response to LPS. Altogether, these results indicate that the global anti-inflammatory activity of MTX relies on its ability to induce a proinflammatory profile in GM-CSF-primed macrophages, making MTX-conditioned macrophages less responsive to proinflammatory stimuli. Our results reconcile the transcriptional17 and functional effects of MTX on human macrophages, and clarify the molecular basis for the anti-inflammatory activity of low-dose MTX.
We also found that TNFAIP3 mRNA expression is significantly higher in PBMCs of MTX-responsive individuals from a cohort of patients with early arthritis under MTX monotherapy. In the context of RA, A20 is a susceptibility gene because (1) polymorphisms of the A20-coding gene TNFAIP3 are associated with RA31–34 and other inflammatory diseases such as systemic lupus erythematosus and psoriasis34; (2) myeloid cell-specific deletion of the Tnfaip3 gene in mice leads to enhanced NF-κB and inflammasome signalling and triggers a spontaneous erosive polyarthritis that resembles human RA35; and (3) A20 mRNA expression in PBMCs is lower in patients with RA compared with healthy individuals.33 The modulation of A20 by MTX that we now report supports the existence of a myeloid-specific effect of the main anchor drug in the treatment of RA and positions A20 as a potential biomarker of responsiveness to MTX, an urgently required parameter to identify those patients with early arthritis who achieve an ACR50 response to MTX monotherapy (usually around 40%).13 Therefore, the analysis of TNFAIP3 polymorphism in MTX-treated patients with RA might be of interest to predict MTX responsiveness in patients with RA and could become a useful MTX response biomarker. Supporting this suggestion, a single nucleotide polymorphism within the OLIG3/TNFAIP3 locus is associated with reduced likelihood to remain on MTX therapy in patients with early RA.36
MTX is considered a prodrug, a compound that undergoes a biochemical modification to become its active form. MTX is polyglutamated once taken up by the cells and MTX polyglutamates (MTX-Pgl) constitute the active form of the drug. MTX-Pgl potently inhibits TS and aminoimidazole-carboxamide-ribonucleoside transformylase. In human macrophages, proinflammatory TS+ GM-CSF-primed macrophages retain higher levels of MTX-Glu2 and MTX-Glu3 than anti-inflammatory TSlow/- M-CSF-polarised macrophages and are more susceptible to MTX.17 Importantly, retention of MTX-Pgl in cells exceeds its half-life in plasma, suggesting that MTX metabolites persist in tissues. In fact, long-lived MTX-Pgl remains in the liver and in bone marrow myeloid precursors for a long period of time.37 The accumulation of MTX in myeloid precursors correlates with the tolerance mechanism that we now describe because macrophages in the inflamed synovial tissue of patients with RA are continuously replaced by circulating monocytes,38 39 and because myeloid precursors appear to contribute to trained innate immunity.40 We hypothesise that monocytes from MTX-treated patients would exhibit impaired responsiveness to danger signals (TNFα, RASF) and that they would display a lower proinflammatory profile than resident macrophages within the inflamed synovia. Therefore, the entry of macrophages with a lower proinflammatory potential into the inflamed synovia would contribute to the beneficial effect of MTX in RA. In any event, the involvement of A20 in MTX response provides a new mechanism of action for MTX, an old drug with low cost and a good safety record.
Acknowledgments
The authors acknowledge Carlota Chiralt and Julia Villarejo for technical help in the sepsis experiments and Dr Angel L. Corbí, Dr Juan D. Cañete and Dr Paloma Sánchez-Mateos for helpful discussions.
References
Footnotes
CM and ÁD-S contributed equally.
Handling editor Josef S Smolen
Contributors CM and AD-S designed research, performed research and analysed data; SF-R, AL, NM, VDC and RGC performed research and analysed data; JLP participated in the research; IG-A designed research and analysed data; AP-K conceived the study, designed research, performed some research, analysed data and wrote the paper. All authors had final approval of the version.
Funding This work was supported by grants from Instituto de Salud Carlos III/FEDER (PI14/00075 and PI17/00037) to AP-K, PI14/00422 to IG-A, RIER RD16 to JLP, IG-A and AP-K, Ministerio de Economía y Competitividad SAF2014-54423-R to ALC. FEDER, Fondo Europeo de Desarrollo Regional: una manera de hacer Europa. SF-R and AP-K are supported by FIBHGM.
Competing interests None declared.
Patient consent Obtained.
Ethics approval This study was approved by Research Ethics Committee of Hospital Universitario La Princesa (PI-518).
Provenance and peer review Not commissioned; externally peer reviewed.