Article Text

Abstract
Objective To evaluate the efficacy and safety of golimumab 50 and 100 mg monotherapy in Japanese patients with active rheumatoid arthritis (RA) despite treatment with disease-modifying antirheumatic drugs (DMARDs).
Methods A total of 316 patients were randomised to receive subcutaneous injections every 4 weeks of placebo (group 1), golimumab 50 mg (group 2) or golimumab 100 mg (group 3); group 1 crossed over to golimumab 50 mg at week 16. The primary end point was the proportion of patients achieving ≥20% improvement in the American College of Rheumatology criteria (ACR20) at week 14. ACR50 and ACR70 response rates were also measured. Adverse events (AEs) were monitored throughout the study.
Results Demographics were similar across groups; the mean age was 52 years and 81.8% of patients (252/308) were female. Week 14 ACR20 response rates were significantly greater in groups 2 (51/101 (50.5%)) and 3 (60/102 (58.8%)) than in group 1 (20/105 (19.0%); p<0.0001 for both), as were ACR50 and ACR70 response rates. After placebo crossover at week 16, week 24 ACR response rates were similar in groups 1 and 2. Through week 16, 63.8% of patients in group 1, 62.4% in group 2 and 60.8% in group 3 had AEs and 1.9%, 1.0% and 2.0% had serious AEs. After week 16, one malignancy was reported (breast cancer, group 3). Infections were the most common AEs. No deaths or cases of tuberculosis were reported through week 24.
Conclusions Golimumab monotherapy (50 and 100 mg) was effective in reducing the signs and symptoms of RA in Japanese patients with active disease despite DMARD treatment.
- Rheumatoid Arthritis
- Anti-TNF
- Treatment
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease characterised by dysregulation of several cytokines, including tumour necrosis factor (TNF).1 ,2 The bone and cartilage damage in the joints can significantly affect physical function3 and the chronic inflammation of RA is associated with significant morbidity and mortality.4 In observational studies, the anti-TNF agents infliximab5 and etanercept6 reduced disease activity in Japanese patients with RA.
Golimumab is a monoclonal antibody that binds with high affinity and specificity to TNF.7 In large, phase 3, randomised, placebo-controlled trials, golimumab demonstrated efficacy in methotrexate (MTX)-naïve8 and MTX-experienced patients with RA.9 In these studies, many patients were treated with concomitant MTX. Some patients cannot tolerate MTX treatment10; therefore, it is clinically relevant to evaluate the safety and efficacy of golimumab monotherapy in Japanese patients with active RA who were previously treated with disease-modifying antirheumatic drugs (DMARDs).
Patients and methods
Patients
Patients (20–75 years) had to have a diagnosis of RA according to the American College of Rheumatology (ACR) criteria11 for ≥3 months and active disease, despite previous DMARD treatment, defined as six or more swollen joints and six or more tender joints and two or more of the following: C-reactive protein (CRP) ≥2.0 mg/dl or erythrocyte sedimentation rate ≥28 mm/h using the Westergren method, morning stiffness ≥30 min, investigator-documented evidence of bone erosion on radiographs, or positive for anti-cyclic citrullinated peptide antibodies or rheumatoid factor. Patients were screened for latent and active tuberculosis (see also online supplementary text). All DMARDs were discontinued ≥4 weeks before the first study agent administration. Concomitant oral corticosteroids (stable dose ≤10 mg of prednisolone/day or equivalent) were permitted.
Study design
This was a phase 2/3 multicentre, randomised, double-blind, placebo-controlled trial carried out at 102 sites in Japan. Patients were randomly assigned (1:1:1) to receive subcutaneous injections every 4 weeks of placebo (group 1), golimumab 50 mg (group 2) or golimumab 100 mg (group 3). Concomitant DMARD treatment, including MTX, was prohibited in all treatment groups (a 4-week washout period was required). At week 16, all patients in group 1 crossed over to receive golimumab 50 mg in a double-blinded fashion.
The study was conducted according to the Declaration of Helsinki and in compliance with good clinical practice guidelines. The protocol was reviewed and approved by the institutional review board at each site. All patients provided written informed consent before any study-related procedures.
Study end points
Response to treatment was evaluated using the ACR criteria, the 28-joint count disease activity score (DAS28) using erythrocyte sedimentation rate and the ACR index of improvement in disease activity (ACR-N); physical function was evaluated with the Health Assessment Questionnaire-Disability Index (HAQ-DI). The primary end point was the proportion of patients achieving ≥20% improvement in ACR criteria (ACR20) at week 14. Due to ethical concerns about the potential for an inadequate response to placebo, week 14 was chosen for the primary efficacy assessment. Secondary end points included ACR50/70/90 response rates at weeks 14 and 24, changes from baseline at weeks 14 and 24 in DAS28 and HAQ-DI scores, ACR-N scores at weeks 14 and 24 and changes from baseline to week 24 in van der Heijde/Sharp (vdH–S) scores. Also the proportions of patients achieving a good or moderate DAS28 score12 ,13 or DAS28 remission (score<2.6) were determined at weeks 14 and 24.
Radiographs of the hands and feet were obtained at baseline and week 24 or at the time of study discontinuation, if applicable, and scored by two independent readers (see online supplementary text). Radiographic progression was evaluated as changes from baseline to week 24 in the vdH–S score.14 Erosion, joint space narrowing and total vdH–S scores are reported. All radiographs were scored by BioClinica Corporation (Newtown, Pennsylvania, USA) and readers were blinded to patient identity, treatment group and time point.
Patients were monitored for adverse events (AEs), including injection-site reactions and abnormal routine laboratory values.
Pharmacokinetic analyses and immunogenicity
Blood samples for the measurement of serum golimumab concentrations were obtained at weeks 0, 4, 8, 12, 14, 16, 20 and 24, with one additional sample between weeks 4 and 12. Blood samples for evaluation of antibodies to golimumab were obtained at weeks 0, 12 and 24. Antibodies to golimumab were detected using a previously described validated antigen bridging enzyme immunoassay.15 Blood samples were drawn before administration of the study agent.
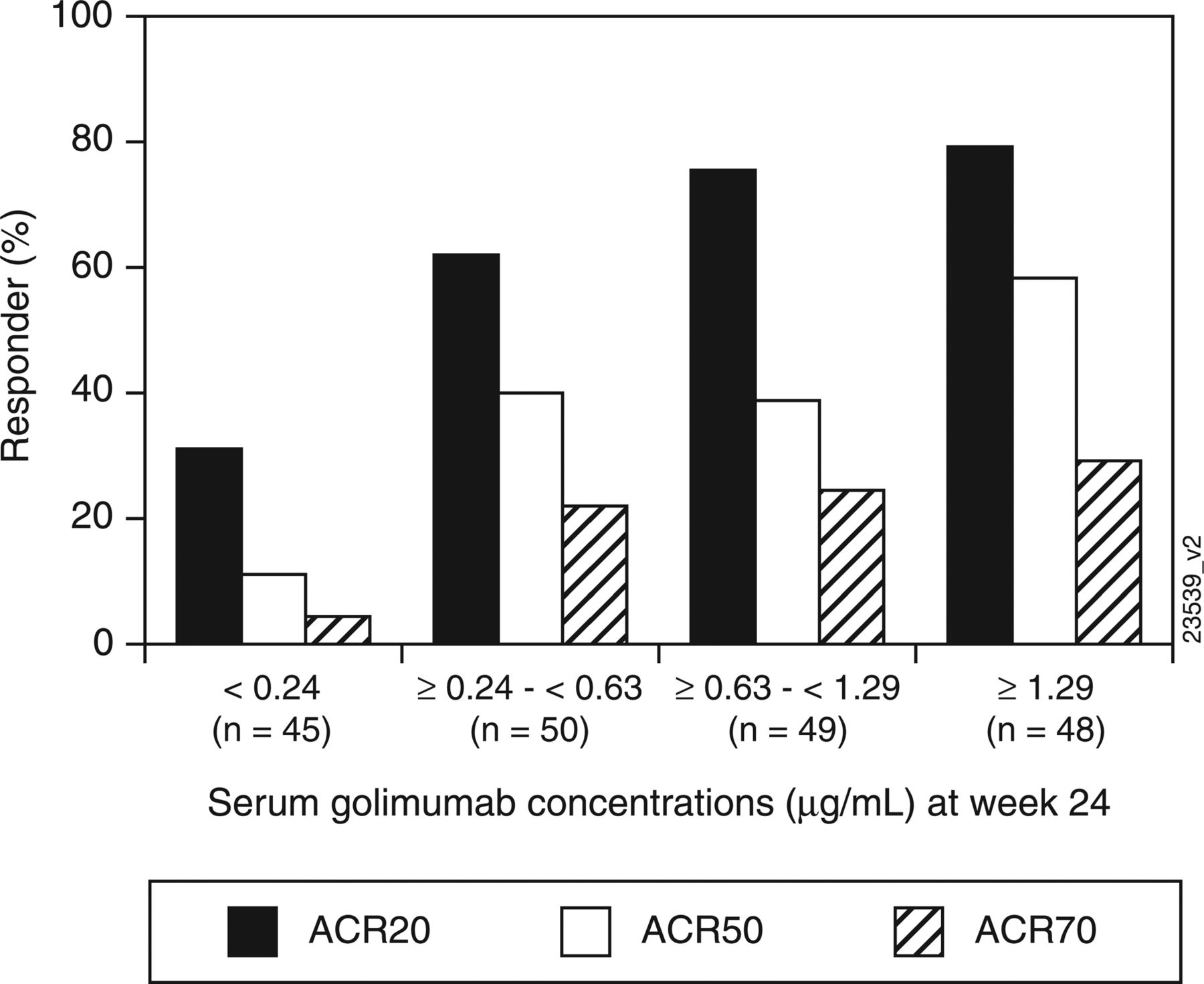
A post hoc analysis evaluated week 24 ACR20, ACR50 and ACR70 response rates for patients stratified according to the following serum golimumab concentration quartiles: <0.24 µg/ml, ≥0.24–<0.63 µg/ml, ≥0.63–<1.29 µg/ml and ≥1.29 µg/ml.
Statistical analyses
All patients who received at least one study agent injection and had efficacy data available were included in the efficacy analysis. All patients who received at least one study agent injection were included in the safety analysis. Patients who received one or more golimumab injection and had pharmacokinetic data available were included in the pharmacokinetic analysis. Descriptive statistics are reported. Differences between the treatment groups in ACR and DAS28 response rates were assessed using a χ2 test. Type I error at the 0.05 level of significance was preserved with a hierarchical approach to control for multiplicity, in which a comparison between groups 3 and 1 was performed first and a comparison between groups 2 and 1 was performed only if the difference between groups 3 and 1 was significant. For changes in continuous variables, treatment group differences were assessed using analysis of covariance (ANCOVA) for HAQ-DI, DAS28 and vdH–S scores or analysis of variance (ANOVA) for ACR-N scores. Least-squares means and 95% CIs are reported. ACR response rates, ACR-N and HAQ-DI were calculated using the last observation carried forward method for the week 14 and week 24 time points. In the analysis of DAS28 response at weeks 14 and 24, observed data were used with no imputation for missing data, with the exception of the DAS28 remission analysis, in which patients with missing data were counted as non-responders. Observed data were used in the pharmacokinetic analysis.
Changes from baseline in vdH–S scores were compared between each golimumab group and placebo using two methods. ANCOVA was the prespecified method in the protocol and was chosen for consistency with the analyses of other continuous variables. A post hoc ANOVA based on van der Waerden normal scores was undertaken to account for the non-normal data distribution due to one patient in group 3 with an atypically large change in vdH–S score. Additionally, a cumulative probability plot of the changes in vdH–S scores from baseline to week 24 for each treatment group was constructed.
Assuming that 5% of patients would be excluded from the efficacy analysis owing to study discontinuation, the target total sample size of 300 patients provided >90% power to detect a difference between groups 2 and 3 and group 1 in ACR20 response rates at week 14 (α=0.05).
Results
Patient disposition and baseline characteristics
A total of 316 patients were randomised; eight withdrew consent before administration of any study agents (figure 1). Therefore, 308 patients received one or more study agent administration (group 1, n=105; group 2, n=101; group 3, n=102). Patient demographics and baseline disease characteristics were well balanced across all groups (table 1). Among all patients, 82% were female, the mean age was 52 years, the mean disease duration was 8.9 years and the mean CRP level was 2.5 mg/dl. Most (73.7%) patients received prior MTX treatment.
Baseline patient demographics and disease characteristics

Patient disposition through week 24. AE, adverse event.
Efficacy results
Clinical response and physical function
At week 14, significantly greater proportions of patients in groups 2 (50.5%) and 3 (58.8%) achieved an ACR20 response in comparison with group 1 (19.0%; p<0.0001 for both) (table 2). Likewise, significantly higher ACR50 and ACR70 response rates were seen in groups 2 and 3 than in group 1. While no patient in group 1 had an ACR90 response at week 14, three patients in group 2 and two in group 3 achieved an ACR90 response; however, statistical significance from placebo was not attained.
Clinical efficacy and radiographic results† through week 24
At week 24, after placebo crossover to golimumab 50 mg at week 16, patients in group 1 generally had ACR response rates similar to those for patients who were initially assigned to group 2 from baseline (table 2). In group 3, week 14 ACR response rates were maintained at week 24.
Mean ACR-N scores at week 14 were significantly greater in groups 2 (30.5) and 3 (33.0) than in group 1 (9.1; p<0.0001 for both) (table 2). Mean improvements from baseline to week 14 in DAS28 scores were also significantly greater in groups 2 and 3 than in group 1 and significantly greater proportions of patients in groups 2 and 3 achieved a moderate or good DAS28 response or DAS28 remission. Improvements from baseline in physical function (HAQ-DI) were also significantly greater in groups 2 and 3 than in group 1.
Patients in group 1 had ACR-N scores at week 24 and mean improvements in DAS28 and HAQ-DI scores from baseline to week 24 that were similar to those seen in patients who were initially randomised to group 2. In group 3, week 14 ACR-N, DAS28 and HAQ-DI responses were maintained at week 24.
Radiographic progression
Two patients did not have complete radiographic data available (missing baseline data for one patient in group 3 and missing week 24 data for one patient in group 2) and changes from baseline in vdH–S score for these patients were substituted with the median change for all patients. Agreement between the two primary readers was good, with intraclass correlation coefficients of 0.98 at baseline and week 24 and 0.80 for the change at week 24. The proportion of patients with a change in total vdH–S score greater than the smallest detectable change was 22.1% (group 1, n=27; group 2, n=21; group 3, n=20).
At week 24, increases in erosion, joint space narrowing and total vdH–S scores were seen in all three groups (table 2), with smaller changes in erosion and total scores in groups 2 and 3, indicating less radiographic progression than in group 1, as shown in the probability plot (figure 2). In the a priori analysis (ANCOVA), no significant differences were seen in mean changes between groups 2 and 3 and group 1 at week 24. In the post hoc ANOVA using normalised scores, no significant differences were seen between groups 2 and 1. Although increases from baseline were observed in both groups 3 and 1, the mean changes in erosion and total vdH–S scores in group 3 were statistically significantly smaller than those in group 1 (1.1 vs 1.3, p=0.0316 and 2.1 vs 2.6, p=0.0043, respectively). Also, the median changes in total vdH–S scores followed a trend, showing less radiographic progression in groups 2 and 3 than in group 1 (0.5 and 0.0, respectively, vs 1.0).
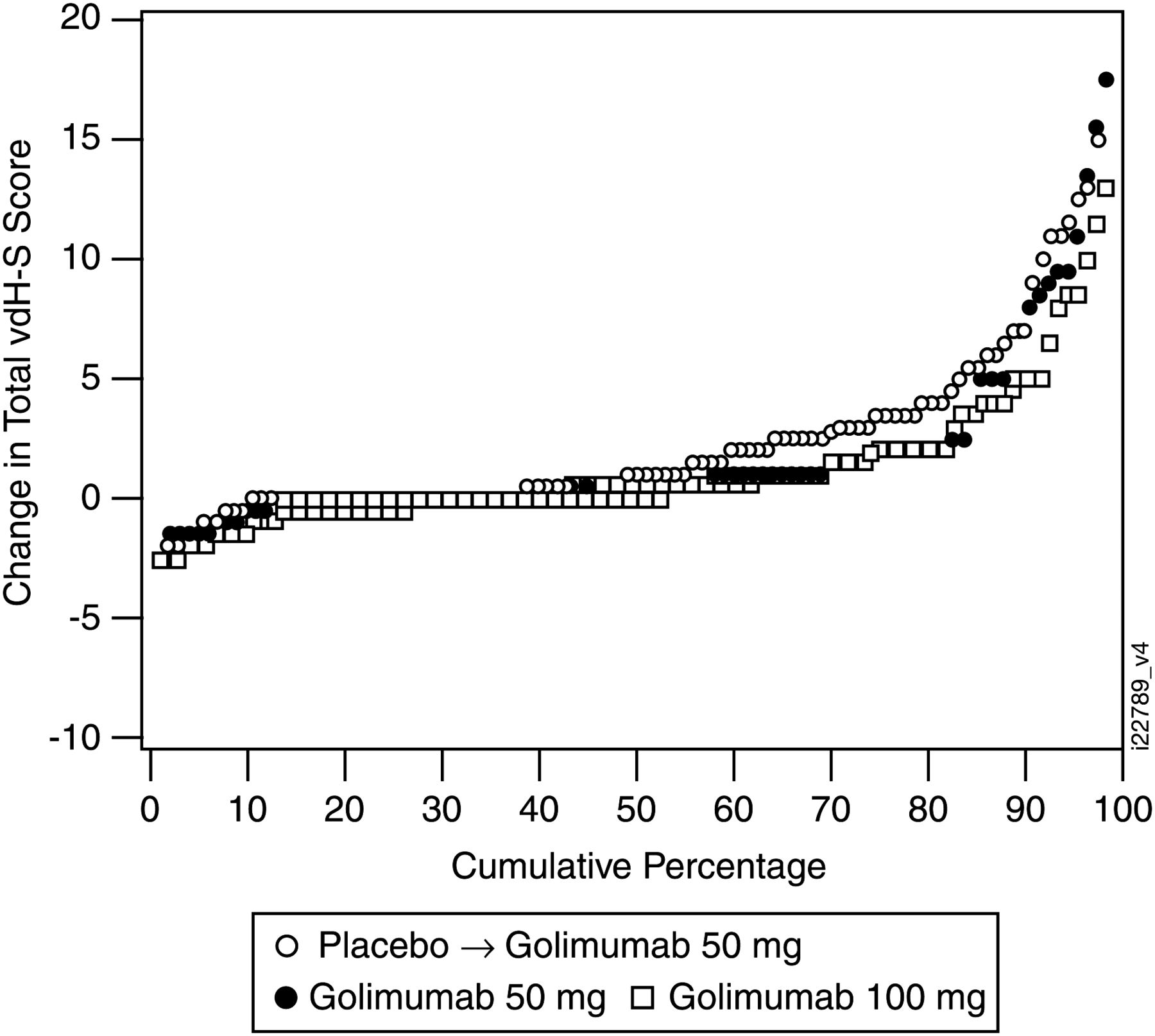
Cumulative probability plot of changes in van der Heijde-Sharp (vdH–S) scores from baseline to week 24 Data from one patient in the golimumab 100 mg group who had an atypically large change in vdH–S score were excluded.
Golimumab pharmacokinetics and antibodies to golimumab
Through week 16, serum golimumab levels increased in a dose-proportional manner; steady state was reached at week 12. Median serum golimumab concentrations for groups 2 and 3, respectively, were 0.52 µg/ml and 1.17 µg/ml at week 12 and 0.46 µg/ml and 1.04 µg/ml at week 16. Median serum concentrations at week 24 were 0.35 µg/ml in group 1, 0.43 µg/ml in group 2 and 0.99 µg/ml in group 3. Week 24 ACR20, ACR50 and ACR70 response rates were evaluated according to serum golimumab concentration, with patients stratified by the following quartiles: <0.24 µg/ml (n=45), ≥0.24–<0.63 µg/ml (n=50), ≥0.63–<1.29 µg/ml (n=49) and ≥1.29 µg/ml (n=48). Overall, response rates were lowest in patients with serum golimumab concentrations <0.24 µg/ml and increased with increasing serum golimumab concentration (figure 3).

{kind=link}
{kind=link}
{kind=link}
The proportions of patients achieving an ACR20, ACR50 and ACR70 responses stratified by serum golimumab concentration quartiles (µg/ml) at week 24. ACR20/50/70, 20%/50%/70% improvement in the ACR criteria.
At week 12, two patients (2.0%) each in groups 2 and 3 tested positive for antibodies to golimumab. At week 24, three patients each in group 1 (3.3%) and group 2 (3.2%) and four patients (4.0%) in group 3 tested positive for antibodies to golimumab. No antibody-positive patient demonstrated an ACR response.
Adverse events
Through week 16 (placebo-controlled period), AEs occurred in 63.8% of patients in group 1, 62.4% in group 2 and 60.8% in group 3 (table 3). Most AEs were mild. The most common AEs were infections (group 1 (23.8%); group 2 (26.7%); group 3 (28.4%)). The most common infections among all golimumab-treated patients were nasopharyngitis (16.3%), pharyngitis (3.4%) and gastroenteritis (2.0%). Three patients (2.9%) in group 1 (herpes zoster, atypical mycobacterial infection and abnormal liver function test), two patients (2.0%) in group 2 (liver disorder and cataract) and one patient (1.0%) in group 3 (transient cerebral ischaemic attack) discontinued the study agent owing to AEs. Serious AEs (SAEs) through week 16 were herpes zoster and organising pneumonia (n=1 each) in group 1, hydrocele (n=1) in group 2 and cellulitis and transient ischaemic attack (n=1 each) in group 3. When assessed by length of follow-up, the incidences (95% CI) of serious infection at week 24 were 3.30 (0.08 to 18.38), 1.69 (0.04 to 9.40) and 2.16 (0.05 to 12.01) for groups 1, 2 and 3, respectively.
Week 16 and week 24 safety results
After the placebo crossover at week 16, AEs occurred in 31 (33.7%) patients in group 1, 34 (35.4%) in group 2 and 33 (33.0%) in group 3 through week 24 (table 3). Infections were the most common AEs during this time period, consistent with results seen during the placebo-controlled period. AEs leading to discontinuation of the study agent after week 16 were ovarian neoplasm (non-malignant; n=1) and RA (n=1) in group 2 and breast cancer (n=1) in group 3. After week 16, SAEs occurred in three patients in group 2 (non-malignant ovarian neoplasm and dental pulpitis, each in one patient; paroxysmal tachycardia and RA in one patient) and in two patients in group 3 (breast cancer, between weeks 20 and 24 and organising pneumonia, one patient each); no SAEs were reported in group 1 during this period.
The incidence of injection-site reactions through week 16 was similar among all groups (group 1, 7/105 (6.7%); group 2, 8/101 (7.9%); group 3, 8/102 (7.8%)). From week 16 through week 24, the rates of injection-site reactions were 3.3% (3/92) in group 1, 6.3% (6/96) in group 2 and 5.0% (5/100) in group 3. All injection-site reactions were mild.
There were no reports of anaphylactic reactions, serum sickness-like reactions, or deaths through week 24. No cases of tuberculosis were reported through week 24; however, one case of atypical mycobacterial infection occurred in group 1 before week 16.
Discussion
In this phase 2/3 study of golimumab 50 mg and 100 mg in Japanese patients with active RA despite DMARD treatment, those treated with golimumab monotherapy had significant improvements from baseline to week 14 in clinical measures of efficacy, including ACR20, ACR50 and ACR70 response rates and DAS28 and ACR-N scores, in comparison with those who received placebo. Physical function was also significantly improved from baseline in the golimumab groups compared with placebo. These significant improvements were seen despite the overall study population displaying relatively mild disease at study outset (mean swollen/tender joint counts of 13/16). However, clinical response to golimumab monotherapy was relatively modest in comparison with golimumab+MTX treatment in another Japanese population.16
Patients with active RA despite previous MTX treatment were evaluated previously in the large phase 3 GO-FORWARD trial.9 While concomitant MTX was included in GO-FORWARD golimumab 100 mg monotherapy was also evaluated. ACR responses were also evaluated at week 14 in both trials and while significantly greater ACR response rates were achieved in group 3 in this study in comparison with placebo, the primary end point was not achieved in the golimumab 100 mg monotherapy group in the GO-FORWARD trial. Possible explanations for the non-statistically significant response in the GO-FORWARD 100 mg monotherapy group were previously described (eg, the relatively low disease activity in the trial population and the high response rate in the MTX monotherapy group).9 However, factors such as patient body weight, which is known to affect the pharmacokinetic properties of monoclonal antibodies,17–19 may also account for the difference in response seen in the two trials. While a previous study found no apparent differences in the pharmacokinetic parameters of golimumab in healthy body-weight-matched Caucasian and Japanese male subjects,20 it is possible that the body weights of patients in 100 mg monotherapy groups in this trial and in GO-FORWARD might have varied considerably21 given that Japanese patients are generally more slight, and the resulting dose per unit mass would be higher than in other populations. Indeed, treatment effects on radiographic progression appear to be related to serum golimumab concentrations, as patients receiving golimumab 50 mg+MTX in the GO-FORTH trial in Japanese patients with RA (week 16 median serum golimumab concentration=0.73 μg/ml) demonstrated significantly less radiographic progression than placebo-treated patients,16 while such a difference was not seen in this study, in which patients receiving golimumab 50 mg had a week 16 median serum golimumab concentration of 0.46 μg/ml.
Radiographic progression was evaluated at week 24, at which point patients randomised to group 1 had been receiving golimumab 50 mg since week 16. The a priori ANCOVA did not show significant differences in radiographic progression between either groups 2 or 3 and group 1; however, in a post hoc analysis using normalised data, significantly smaller changes from baseline in erosion and total vdH–S scores were seen in group 3 than in group 1. This significant difference was confirmed by an additional ANCOVA that excluded a single group 3 patient with an atypically large change in vdH–S score (p=0.01; data not shown). Biological monotherapy with the anti-interleukin 6 agent tocilizumab has also demonstrated radiographic benefit in patients with RA with inadequate response to DMARD treatment.22 In this study, the mean baseline CRP level, which is a good predictor of radiographic progression,23 was moderately raised and 22.1% of patients had a change in total vdH–S that exceeded the smallest detectable change. In contrast, only 4.3% of patients in GO-FORWARD had such a change in total vdH–S score.24 Thus, patients in our study probably had higher disease activity than patients in GO-FORWARD. This may account for the observation that radiographic progression in this study was greater than expected based on the clinical response seen at similar time points in earlier golimumab trials, including GO-FORWARD.24 Our results suggest that golimumab 100 mg monotherapy may prevent further joint damage in Japanese patients with active radiographic progression, which is consistent with the golimumab package insert approved by the Japanese Pharmaceuticals and Medical Devices Agency.25
Golimumab was generally well tolerated. Infections were the most common AEs. Serious infections were reported in two patients through week 16 and one patient between weeks 16 and 24; the week 24 incidences per 100 patient-years of follow-up indicated no increase in serious infection versus placebo. Most AEs were mild and few patients discontinued due to AEs. Rates of SAEs, serious infections and malignancies were low. No deaths and one malignancy (breast cancer) occurred through week 24. Of note, this study was not powered to detect rare events and these findings are limited also by the short-term nature of the analysis.
This was the first golimumab monotherapy study to demonstrate that Japanese patients with active RA despite prior DMARD treatment had significantly improved signs and symptoms of RA after 14 weeks of treatment with 50 or 100 mg golimumab in comparison with placebo. Group 3 had significantly less radiographic progression than group 1 when analysed post hoc using normalised scores, and median changes in total vdH–S scores suggested a dose-dependent trend. Additional long-term analyses are needed to further explore the effect of golimumab monotherapy on joint destruction and fully assess its safety profile in Japanese patients with RA.
Acknowledgments
We thank the patients, investigators and study personnel who made this trial possible. We also thank Rebecca Clemente, PhD, Michelle Perate, MS, and Mary Whitman, PhD, (Janssen Services, LLC); Yoshifumi Ukyo and Yoshinori Murakami, PhD (Janssen Pharmaceutical K.K) for assistance in preparing this manuscript.
References
Supplementary materials
Supplementary Data
Files in this Data Supplement:
Footnotes
Handling editor Tore K Kvien
-
Contributors All authors contributed to the design and/or conduct of the trial, analysis and/or interpretation of data and manuscript preparation and/or review for critical content. All authors also approved the final manuscript for submission to ARD. RC, MP and MW (Janssen Services, LLC) and YU and YM (Janssen Pharmaceutical K.K) provided assistance with preparing this manuscript.
-
Funding This study was funded by Janssen Pharmaceuticals KK
-
Competing interests DB is an employee of Janssen Research & Development, LLC and owns stock in Johnson & Johnson. HY has received research grants from Abbott, Bristol Myers Squibb, Chugai Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharmaceutical, Otsuka Pharmaceutical, Roche, Takeda Pharmaceutical and Wyeth. KY has received research grants from Astellas Pharmaceutical, Chugai Pharmaceutical, Eizai Pharmaceutical, Immunofuture Inc, Mitsubishi Tanabe Pharma Corporation, Santen Pharmaceutical and Wyeth. MH has received research grants from Abbott, Bristol Myers Squibb, Chugai Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharma Corporation, Takeda Pharmaceutical and Wyeth and received consultant fees from Abbott, Bristol Myers Squibb, Chugai Pharmaceutical, Janssen Pharmaceutical and Mitsubishi Tanabe Pharma Corporation. MK has received research grants from Astellas Pharmaceutical, Astra Zeneca, Banyu Pharmaceutical, Daiichi Sankyo Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, GlaxoSmithKline, Mitsubishi Tanabe Pharma Corporation, Nippon Boehringer Ingelheim and Novartis. NI has received research grants from Astellas Pharmaceutical, Chugai Pharmaceutical, Eizai Pharmaceutical and Mitsubishi Tanabe Pharma Corporation. NM has received research grants from Abbott, Astellas Pharmaceutical, Banyu Pharmaceutical, Chugai Pharmaceutical, Daiichi Sankyo Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharma Corporation, Takeda Pharmaceutical and Teijin Pharmaceutical. TK has received research grants from Abbott, Bristol Myers Squibb, Chugai Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharma Corporation, Otsuka Pharmaceutical, Pfizer, Takeda Pharmaceutical and Wyeth. TO is an employee of Janssen Pharmaceutical KK, a wholly owned subsidiary of Johnson & Johnson. TT has received research grants from Abbott, Astra Zeneca, Bristol Myers Squibb, Chugai Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharma Corporation, Novartis, Takeda Pharmaceutical and Wyeth. TY is an employee of Mitsubishi Tanabe Pharma Corporation. YT has received research grants from Abbott, Astellas Pharmaceutical, Banyu Pharmaceutical, Chugai Pharmaceutical, Eizai Pharmaceutical, Janssen Pharmaceutical, Mitsubishi Tanabe Pharma Corporation, Pfizer and Takeda Pharmaceutical.
-
Provenance and peer review Not commissioned; externally peer reviewed.