Article Text

Abstract
Objective To assess the efficacy and safety of low-dose prednisone chronotherapy using a new modified-release (MR) formulation for the treatment of rheumatoid arthritis (RA).
Methods In this 12-week, double-blind, placebo-controlled study, patients with active RA (n=350) were randomised 2:1 to receive MR prednisone 5 mg or placebo once daily in the evening in addition to their existing RA disease-modifying antirheumatic drug (DMARD) treatment. The primary end point was the percentage of patients achieving a 20% improvement in RA signs and symptoms according to American College of Rheumatology criteria (ie, an ACR20 response) at week 12. Changes in morning pain, duration of morning stiffness, 28-joint Disease Activity Score and health-related quality of life were also assessed.
Results MR prednisone plus DMARD treatment produced higher response rates for ACR20 (48% vs 29%, p<0.001) and ACR50 (22% vs 10%, p<0.006) and a greater median relative reduction from baseline in morning stiffness (55% vs 35%, p<0.002) at week 12 than placebo plus DMARD treatment. Significantly greater reductions in severity of RA (Disease Activity Score 28) (p<0.001) and fatigue (Functional Assessment of Chronic Illness Therapy-Fatigue score) (p=0.003) as well as a greater improvement in physical function (36-item Short-Form Health Survey score) (p<0.001) were seen at week 12 for MR prednisone versus placebo. The incidence of adverse events was similar for MR prednisone (43%) and placebo (49%).
Conclusion Low-dose MR prednisone added to existing DMARD treatment produced rapid and relevant improvements in RA signs and symptoms.
ClinicalTrials.gov, number NCT00650078
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Glucocorticoids such as prednisone are established components of treatment strategies for many inflammatory conditions, such as rheumatoid arthritis (RA), and are widely used.1,–,3 Accumulating evidence suggests that low-dose treatment is well tolerated and minimises the risk of the undesirable effects associated with higher doses.4 However, there is still a need to improve the risk–benefit profile for these valuable anti-inflammatory drugs by increasing the efficacy of low-dose treatment. One promising approach is chronotherapy, in which the delivery of treatment is coordinated with circadian biological rhythms. The chronotherapeutic approach has shown promise in several therapeutic areas, including the management of hypertension, allergic rhinitis and bronchial asthma.5,–,7
Chronotherapy may be particularly appropriate for RA because symptoms follow circadian rhythms, with impaired function due to pain and joint stiffness commonly being most severe in the early morning.8 ,9 Emergence of these symptoms follows the increase in serum levels of interleukin 6 (IL-6; a key inflammatory mediator), tumour necrosis factor α (TNFα) and other proinflammatory cytokines that occur late at night.9,–,13 Nocturnal secretion of cortisol, which can counter the effects of increased IL-6 levels, is also perturbed in patients with RA and may contribute to the emergence of morning symptoms.8 ,14 These observations suggest that the optimal time for delivery of glucocorticoid treatment is during the night, to mimic the normal circadian rhythm of cortisol secretion and target the effects of nocturnal proinflammatory stimuli.
A modified-release (MR) formulation of prednisone has been developed to deliver prednisone chronotherapy. This innovative tablet uses a programmed-release mechanism to release prednisone approximately 4 h after ingestion (ie, at approximately 02:00 am if the patient takes the tablet at 10:00 pm). We report the results of a double-blind, placebo-controlled, multicentre study (Circadian Administration of Prednisone in Rheumatoid Arthritis, CAPRA-2) that investigated the efficacy and safety of low-dose prednisone chronotherapy in patients with active RA.
This is the first rigorous placebo-controlled study to investigate the efficacy of low-dose prednisone in patients with active disease receiving disease-modifying antirheumatic drug (DMARD) treatment and according to current standards. It thus allows comparison with the results of recent studies of other treatments in patients with active RA.
Methods
Study design
In this 12-week, double-blind, parallel-group, placebo-controlled study, following a 1-week screening period, eligible patients were randomised 2:1 to receive MR prednisone (5 mg) or placebo once daily, taken with or after their evening meal, in addition to their standard RA treatment.
The study was conducted in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice and the Declaration of Helsinki. The protocol was approved by the ethics committees and institutional review boards of all centres, and all patients provided written informed consent before study-related procedures. The trial is registered at ClinicalTrials.gov, number NCT00650078.
Patients
Patients aged 18–80 years with a diagnosis and documented history of RA and who had been taking DMARDs for at least 6 months were eligible for inclusion. Patients were also required to have had a duration of morning stiffness of at least 45 min on at least 4 days within the 7 days of screening, a swollen joint count of ≥4 and a tender joint count of ≥4. Patients receiving oral glucocorticoids within 6 weeks of the screening visit were excluded from the study (see online supplementary material for further details). The study protocol prohibited initiation of any new DMARD or non-steroidal anti-inflammatory drug (NSAID) treatment during the study; changes to existing DMARD treatment (dosing and frequency) were also prohibited.
Outcomes and follow-up
Scheduled study visits occurred at baseline and weeks 2, 6 and 12, and were to occur between 08:00 and 10:00 pm. At each visit, doctors assessed the number of tender and swollen joints and global disease activity, and patients assessed pain and global disease activity and completed the Functional Disability Index of the Health Assessment Questionnaire.15 Disease activity at each visit was determined using the 28-joint Disease Activity Score (DAS28).16 Assessments of pain and global disease activity were made using 0–100 mm visual analogue scales (0=no pain/not active at all; 100=very intense pain/extremely active). Blood samples were collected at each study visit.
Throughout the study, patients completed a diary card twice daily. In the mornings they recorded whether they had joint stiffness and its severity, the time of resolution of joint stiffness and pain levels on waking. Evening assessments included pain intensity during the day and whether the patient had experienced recurrence of stiffness. Patients assessed their health status using the 36-item Short-Form Health Survey (SF-36),17 ,18 and the Functional Assessment of Chronic Illness Therapy-Fatigue (FACIT-F) questionnaire19 ,20 at baseline and week 12. Safety assessments (recording of adverse events (AEs) and vital signs) were performed at each study visit according to standard procedure (ie, without using checklists with predefined events).
The primary efficacy end point was the proportion of patients with a 20% improvement in RA signs and symptoms according to American College of Rheumatology (ACR) criteria (ie, an ACR20 response)21 at week 12. A key secondary end point was the change in duration of morning stiffness between baseline and week 12. (See online supplementary material for details of secondary end points.)
Statistical analysis
The study aimed to demonstrate a difference of at least 20% in ACR20 for MR prednisone versus placebo at week 12. The sample size calculation was based on comparison of two proportions using the χ2 test and a randomisation ratio of 2:1 for MR prednisone:placebo. Assuming an ACR20 response rate of 25% for placebo, 294 patients would be required to provide 90% power to detect an ACR20 response rate of 45% in the MR prednisone group at a significance level of α=0.05. The study therefore aimed to randomise at least 294 patients; in order to account for potential drop-outs, a total of 350 patients were recruited to the study.
Duration of morning stiffness was the difference between the time of resolution of morning stiffness and the time of waking. The difference between the treatment groups was assessed using the median and the 95% CI of the median, computed using the Hodges–Lehmann method. (See supplementary material for further details.)
Results
Study population
A total of 350 patients were randomised between April 2008 and February 2009; of these, 323 (92.3%) completed the study (figure 1 and supplementary table 1). The main reasons for early withdrawal were AEs and patient requests. Demographics and baseline disease characteristics were generally well balanced between the two treatment groups (table 1). The study population was primarily female (84%), aged >45 to <65 years (70%), and about half of the study population (55%) had had RA for at least 5 years. All patients had previously received treatment for RA: 99% with DMARDs and 73% with NSAIDs.

Patient disposition. A total of 350 patients were enrolled from 50 centres in six countries: Germany (3 centres, 3 patients), UK (3 centres, 12 patients), Poland (10 centres, 145 patients), Hungary (9 centres, 102 patients), Canada (2 centres, 13 patients) and USA (23 centres, 75 patients). AE, adverse event; MR, modified release.
Demographics and baseline disease characteristics
Virtually all patients (>98%) received concomitant DMARD treatment, the most frequently used being methotrexate (73.7% of patients), sulfasalazine (14.6%) and leflunomide (11.1%). Analgesic use was similar between treatment groups (MR prednisone, 83.1%; placebo, 86.6%). The most frequently used analgesics were anilides (27.4% of patients), acetic acid derivatives (25.1%) and propionic acid derivatives (17.1%). Detailed analysis showed no significant changes in DMARD and NSAID use between baseline and end of study (supplementary table 2) indicating that observed results were not confounded by changes in concomitant treatment.
Efficacy
ACR response rate
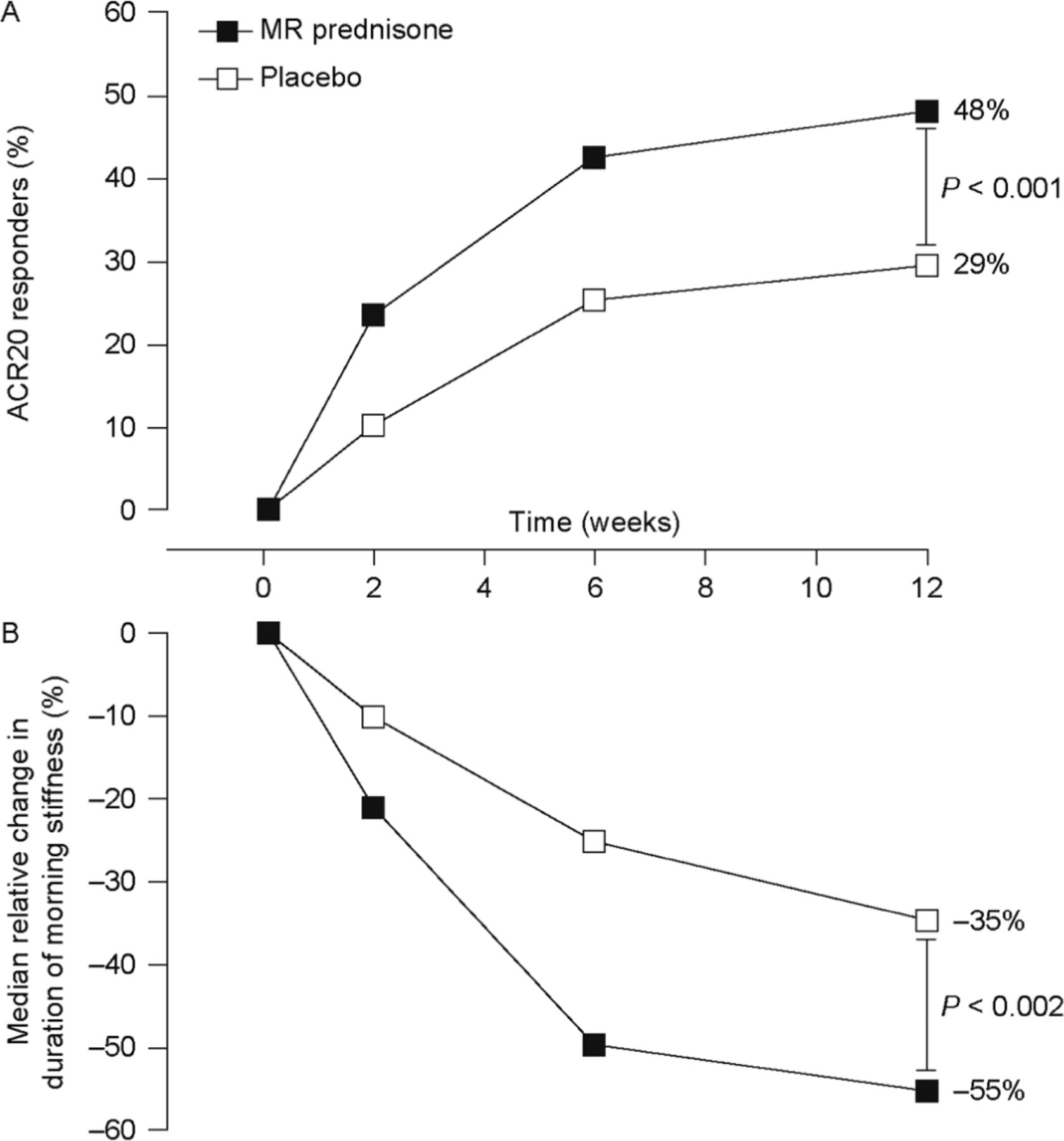
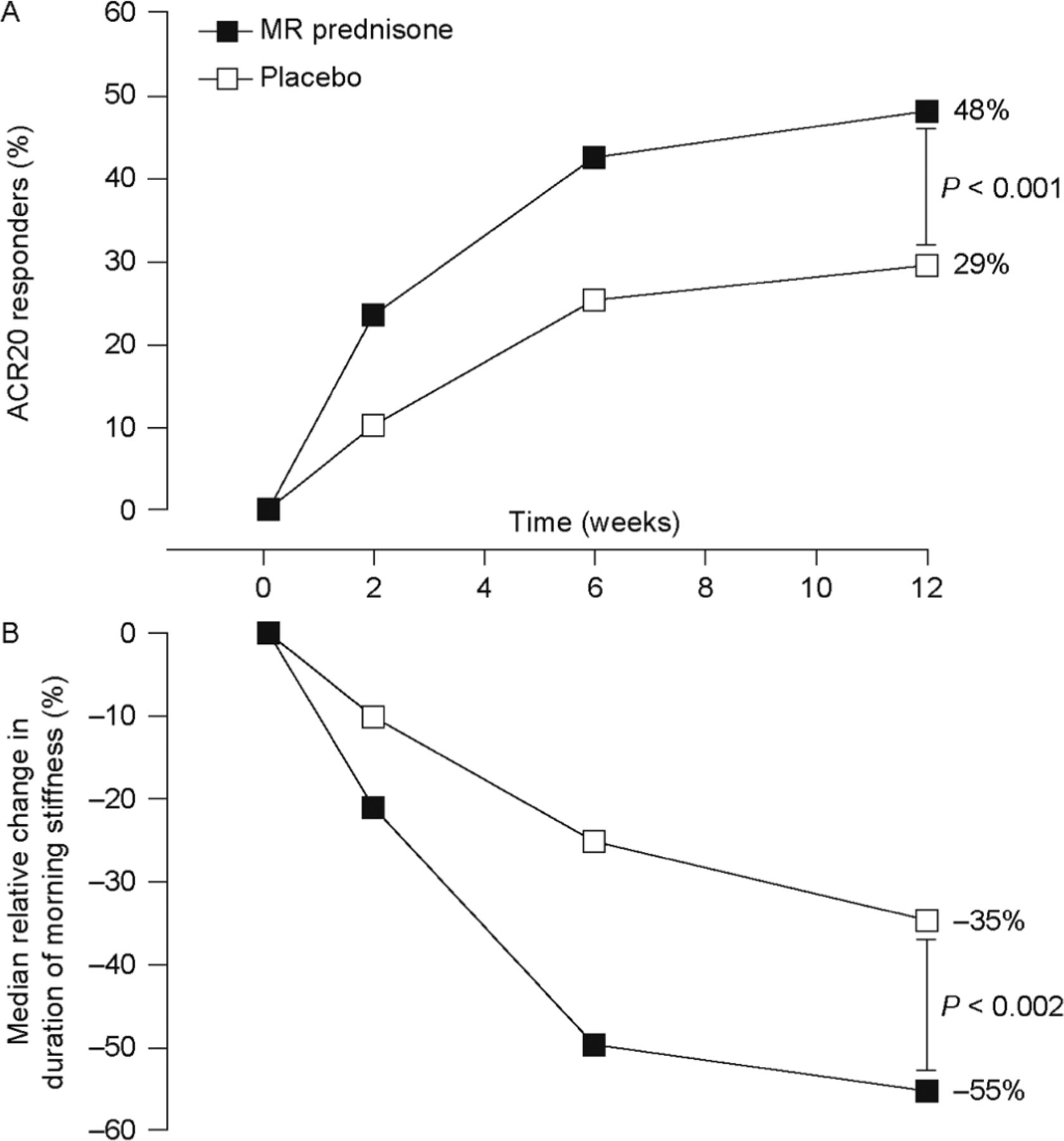
ACR20 and ACR50 response rates at week 12 were significantly greater with MR prednisone than with placebo. At week 12, 48% of patients receiving MR prednisone achieved an ACR20 response, compared with 29% in the placebo group, a difference of 19% (p<0.001). The response was achieved rapidly: a significant difference in ACR20 response rate between treatment groups was evident at week 2, and the difference remained significant throughout the study (p<0.005) (figure 2A).
{kind=link}
{kind=link}
Improvements in rheumatoid arthritis symptoms. (A) Percentage of patients achieving a 20% improvement in rheumatoid arthritis signs and symptoms according to American College of Rheumatology criteria (ACR20) (primary end point). p<0.003 for the between-group difference at weeks 2, 6 and 12. (n Values for weeks 2, 6 and 12 were 231, 229 and 229, respectively, for the modified-release (MR) prednisone group and 119, 119 and 119, respectively, for the placebo group.) (B) Change in duration of morning stiffness from baseline. p<0.004 for the between-group difference at weeks 2, 6 and 12. (n Values for weeks 2, 6 and 12 were 228, 220 and 216, respectively, for the MR prednisone group and 119, 112 and 107 for the placebo group.)
ACR50 responder rates were numerically greater with MR prednisone than with placebo at all time points, and the difference was significant at weeks 6 and 12 (22% vs 10% at week 12, p<0.006). Few patients had an ACR70 response at week 12: 7% of those taking MR prednisone and 3% of placebo recipients (p=0.10).
Individual ACR core set measures
All individual ACR core set measures except C-reactive protein and erythrocyte sedimentation rate showed significantly greater improvements from baseline to week 12 with MR prednisone than with placebo (table 2). Changes from baseline were also significantly different between the placebo and MR prednisone groups at weeks 2 and 6 for all clinical end points (p<0.05).
Mean change from baseline at week 12 in clinical variables and health-related quality-of-life end points*†
Patients achieving low disease activity
MR prednisone significantly increased the proportion of patients achieving low disease activity (defined as having a 28-joint Disease Activity Score (DAS28)≤3.2) after 6 weeks (p<0.001) and 12 weeks (p=0.0109) of treatment (supplementary table 3). At 12 weeks, 11.3% patients in the MR prednisone group had achieved a DAS28 score <2.6 (disease remission or, according to Felson et al,22 minimal disease activity) compared with 6.7% in the placebo group.
Morning stiffness
At baseline, the median duration of morning stiffness was similar between the two treatment groups: MR prednisone, 127 min; placebo, 139 min. At week 12, the median duration of morning stiffness was 46 min in the MR prednisone group (median relative reduction from baseline of 55%), compared with 79 min for placebo (median relative reduction from baseline of 35%). The difference between groups in median relative reduction in duration of morning stiffness was significant at weeks 2, 6 and 12 (p<0.004 for all comparisons) (figure 2B). Significantly greater decreases in the severity of morning stiffness and recurrence of stiffness later in the day were also seen for MR prednisone compared with placebo (p≤0.01) (table 2). Further analysis showed no correlation between disease duration and effect on morning stiffness (supplementary table 4) and a regression analysis showed that duration of RA was not a predictor of reduced duration of morning stiffness (p=0.8433).
Morning and evening pain
At baseline, both groups reported having considerable morning pain (table 1). Reductions in morning pain from baseline were seen in both treatment groups and were significantly greater in the MR prednisone group at all time points (p≤0.05) (table 2). Significantly greater reductions in evening pain from baseline were also seen for the MR prednisone group (p<0.05) (table 2).
Health-related quality of life
At baseline, patients were experiencing considerable fatigue compared with the general population, as indicated by mean FACIT-F scores (MR prednisone, 29; placebo, 29; general population, 44).19 FACIT-F scores increased in both treatment groups over the course of the study, indicating a reduction in fatigue; the change was significantly greater in the MR prednisone group (p=0.003) (table 2).
Improvements in physical function and mental function were also observed over the course of the study in both treatment groups, according to SF-36 assessments (table 2). At baseline, mean scores for physical function (MR prednisone, 32; placebo, 31) were well below that of the US general population—namely, 50.18 The improvement in physical function was significantly greater in the MR prednisone group (3.6 vs 1.3, p<0.001).
Laboratory variables
IL-6 levels at screening were highly variable (table 1), and more than 50% of patients had levels below the limit of detection. Over the 12-week study, IL-6 levels decreased in both treatment groups. The decrease in IL-6 was greater in the MR prednisone group as evident from the geometric mean titre ratio of 0.8 (95% CI 0.7 to 0.9). Minor increases in C-reactive protein levels and decreases in erythrocyte sedimentation rate were seen over the course of the study and were similar in the two treatment groups (table 2). TNFα levels in the two groups were comparable at baseline (table 1) and levels remained unchanged over the 12-week study; the geometric mean titre ratio was 1.0 (95% CI 0.97 to 1.04) for the change in TNFα levels between treatments.
Safety and tolerability
MR prednisone was generally well tolerated, and there were no deaths or life-threatening AEs. The incidence of AEs was slightly lower in the MR prednisone group than the placebo group (43% vs 49%). The incidence of AEs regarded by investigators as being related to treatment was similar in the two groups (7.8% vs 8.4%) (table 3). In both treatment groups, the most frequently occurring AEs were related to worsening of the underlying disease—namely, arthralgia and aggravated RA/RA flare-up, and these occurred more frequently in the placebo group. The difference in incidence was statistically significant for arthralgia (p=0.0141), but not for aggravated RA/RA flare-up (p=0.3917). The incidence of infections was similar for the two groups (MR prednisone, 13%; placebo, 12%), as was the incidence of the most frequently reported infection, nasopharyngitis; bronchitis was reported more frequently for the placebo group, though the increase was not significant (table 3). Most events were mild or moderately severe.
Adverse events
Serious AEs were reported for one patient (0.4%) receiving MR prednisone and two (1.7%) receiving placebo (table 3); none of the serious AEs were considered severe or related to study treatment. Six patients withdrew from the study because of AEs, five (2.2%) in the MR prednisone group (due in one case each to: headache, headache and hypertension, glaucoma, RA flare and vomiting) and one (0.8%) in the placebo group (due to headache); all events except the case of RA flare in the MR prednisone group were considered related to treatment. No clinically relevant changes in haematological or biochemical parameters or vital signs were seen during the study.
Discussion
Low-dose MR prednisone chronotherapy has an important clinical effect on symptoms of RA in patients with active disease receiving conventional DMARDs, as evident from ACR20 and ACR50 response rates at week 12 in this study. Clinical responses were achieved rapidly, with most clinical end points showing statistically significant differences for MR prednisone over placebo as early as 2 weeks after the start of treatment, and responses were maintained for the duration of the 12-week study. Significant improvements in health-related quality of life (HRQoL) were also seen. The use of analgesics was similar between groups, indicating that the observed improvements could be attributed to MR prednisone, rather than differences in analgesic medication.
Prednisone chronotherapy is expected to have a particular impact on morning symptoms of RA. This is borne out in this study, in which MR prednisone reduced the duration of morning stiffness from approximately 2 h at baseline to 46 min at week 12, a median relative reduction of 55% which was approximately 1.5-fold greater than that seen with placebo. Morning pain, severity of morning stiffness and RA severity (according to DAS28 score) were also considerably reduced with MR prednisone over the 12-week study. This is in agreement with results from our previous study (CAPRA-1),23 in which MR prednisone induced greater improvements in morning stiffness and reductions in IL-6 levels than immediate-release (IR) prednisone. These results suggest that the timing of delivery significantly affects the efficacy of glucocorticoid treatment and that chronotherapy may allow efficacious treatment with lower glucocorticoid doses.
Previous studies of low-dose prednisone have largely investigated the benefits of adding low-dose (≤10 mg/day) IR prednisone to DMARDs in patients with early RA.25,–,30 These placebo- controlled studies have demonstrated more rapid improvements in clinical symptoms over the first 6 months of treatment for prednisone compared with placebo, and are thus in agreement with our results obtained in patients with more advanced disease.25 ,28,–,30 While numerical differences in favour of the prednisone group were also evident at 12 or 24 months, differences were no longer statistically significant in most cases.25,–,28 However, the addition of low-dose prednisone has been reported to increase the probability of achieving remission over the first year of treatment and of maintaining remission beyond the first year,30 and to decrease radiographic progression.25 ,27,–,29 Given the similar results reported for IR prednisone and MR prednisone over the first months of treatment, prolonged treatment with MR prednisone can also be expected to slow radiographic progression, but this disease-modifying effect has still to be proved.
We report that MR prednisone was well tolerated. In this 12-week study, the overall incidence of AEs was slightly lower in patients receiving MR prednisone than in those receiving placebo, and none of the serious or severe AEs in the MR prednisone group was considered related to treatment. In addition, there was no evidence for an increased risk of infection with active treatment; indeed the incidence of bronchitis was higher in the placebo group. The incidences of hypertension and discontinuation due to AEs were low but were slightly higher in the MR prednisone group. Notably, the incidences of arthritis and arthralgia reported as AEs were higher in the placebo group, again reflecting the efficacy of MR prednisone. The safety profile of MR prednisone presented here is similar to that seen in the CAPRA-1 study11 ,23 and in placebo-controlled studies for IR prednisone.27 ,29
Our study has several limitations. First, patients were required to have morning stiffness of more than 45 min to be included in the study; our results may thus not be directly applicable to patients with less severe disease. Second, this was a 12-week study. This duration is sufficient to demonstrate the initial benefits achieved by adding MR prednisone to DMARD treatment, including improvements in morning function and HRQoL. However, the study did not assess effects on structural damage and disease progression, which would require longer follow-up. Third, while the results of this study demonstrate that short-term treatment with MR prednisone has a similar safety profile to that of placebo, long-term studies are required to assess the safety and tolerability of prolonged treatment.
In fact, this has already been demonstrated in the open-label extension to the CAPRA-1 study, where patients received either MR prednisone or IR prednisone for 3 months, before receiving MR prednisone for 9 months. The only AEs reported in >2% of patients during the 9-month extension (months 4–12) were RA-related symptoms (14.5%), upper respiratory tract infections (2.8%), back pain (2.8%) and weight increase (2.8%).11 An integrated safety analysis (supplementary tables 5 and 6) provides further safety data from the full 12 months of CAPRA-1 (either 12 months MR prednisone treatment or 3 months IR prednisone treatment (months 0–3) followed by 9 months MR prednisone treatment (months 4–12) depending on initial randomisation) and combined safety data for patients receiving MR prednisone for 3 months from both CAPRA-1 and CAPRA-2.31 The incidence of AEs was higher over the 12-month period than for the 3-month period (as would be expected for the longer duration of treatment), though the increase was not proportional to the duration of treatment. For example, the incidence of severe AEs during the first 3 months of treatment was 2.4% (supplementary table 5) compared with 3.3% in patients receiving 12 months MR prednisone treatment (supplementary table 6). Similarly, aggravated RA/RA flare-up was reported in 12.8% of patients during the first 3 months and in 14.2% of patients during the 12-month treatment period (supplementary tables 5 and 6). The only AEs reported in ≥4% of patients receiving MR prednisone for 12 months were aggravated RA/RA flare-up and flushing (supplementary table 6).
In conclusion, the results of this study demonstrate that even at a dose considered to be below substitution levels, MR prednisone chronotherapy is highly effective and well tolerated in patients with RA, providing rapid relief of symptoms and, particularly, improving morning function. Further, longer-term studies are warranted to determine the dose and strategy that optimises the benefit-to-risk ratio for MR prednisone in the management of RA.
Acknowledgments
The authors thank the patients and investigators who took part in this study. The authors take full responsibility for the content of the paper. They thank Patricia Rice, MS (CliniRx USA) and Sandrine Cayez, MS (ICON Clinical Research SARL) for statistical analysis of the data; Rowena Hughes, PhD and Adam Giles, PhD (Oxford PharmaGenesisTM) for medical writing support, editorial assistance and collation and incorporation of comments from all authors; Amy Grahn, MS (Clinical Development and Operations, Horizon Pharma) for thorough data review, and Ivonne Mitar, DPhil (Medical Science Liaison, Horizon Pharma, formerly Nitec Pharma GmbH) for critical review and for coordinating the writing of the manuscript. The editorial help was supported by Horizon Pharma, Mannheim, Germany and Northbrook, IL, USA.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
-
Funding Horizon Pharma (formerly Nitec Pharma), Mannheim, Germany and Northbrook, Illinois, USA.
-
Competing interests FB received consultancy fees, honoraria and travel expenses from Merck Serono, Horizon Pharma (formerly Nitec Pharma) Mundipharma Int Ltd and grant support from Merck Serono and Horizon Pharma. JK received honoraria, consultancy fees, grants and travel expenses paid to his institution from Horizon Pharma (formerly Nitec Pharma), AstraZeneca, CombinatoRx, GlaxoSmithKline, Merck and Wyeth. MB received consultancy fees from Augurex, Bristol-Myers Squibb, CombinatoRx, GlaxoSmithKline, Medimmune, Horizon Pharma (formerly Nitec Pharma), Mundipharma and Roche; honoraria from Genentech, Novartis and Sanofi; and payment for development of educational presentations from Schering-Plough and UCB. REA received consultancy fees and honoraria from Merck Serono and Horizon Pharma (formerly Nitec Pharma) and travel expenses and payment for development of educational presentations from Merck Serono. KGS received consultancy fees, honoraria and travel expenses from Merck Serono, Horizon Pharma (formerly Nitec Pharma), Novartis, Roche, Amgen, UCB, Genentech and Eli Lilly, and grant support from Merck Serono, Proctor & Gamble, Roche, GlaxoSmithKline, Amgen and Eli Lilly. SW and UR are employees of Horizon Pharma and have stock options. DM, JSz, JSu and IS reported no conflicts of interest.
-
Provenance and peer review Not commissioned; externally peer reviewed.