Article Text

Abstract
Objective A number of rheumatoid arthritis (RA) susceptibility genes have been identified in recent years. Given the overlap in phenotypic expression of synovial joint inflammation between RA and psoriatic arthritis (PsA), the authors explored whether RA susceptibility genes are also associated with PsA.
Methods 56 single nucleotide polymorphisms (SNPs) mapping to 41 genes previously reported as RA susceptibility loci were selected for investigation. PsA was defined as an inflammatory arthritis associated with psoriasis and subjects were recruited from the UK and Ireland. Genotyping was performed using the Sequenom MassArray platform and frequencies compared with data derived from large UK control collections.
Results Significant evidence for association with susceptibility to PsA was found toa SNP mapping to the REL (rs13017599, ptrend=5.2×104) gene, while nominal evidence for association (ptrend<0.05) was found to seven other loci including PLCL2 (rs4535211, p=1.7×10−3); STAT4 (rs10181656, p=3.0×10−3) and the AFF3, CD28, CCL21, IL2 and KIF5A loci. Interestingly, three SNPs demonstrated opposite effects to those reported for RA.
Conclusions The REL gene, a key modulator of the NFκB pathway, is associated with PsA but the allele conferring risk to RA is protective in PsA suggesting that there are fundamental differences in the aetiological mechanisms underlying these two types of inflammatory arthritis.
This paper is freely available online under the BMJ Journals unlocked scheme, see http://ard.bmj.com/info/unlocked.dtl
Statistics from Altmetric.com
Introduction
Psoriatic arthritis (PsA) shares many features in common with rheumatoid arthritis (RA). For example, both diseases are characterised by the occurrence of an inflammatory arthritis in peripheral synovial joints; both respond to similar therapies including Methotrexate and anti-tumour necrosis factor biologic treatment and both are complex diseases with genetic and environmental components to susceptibility. Much progress has been made in identifying RA susceptibility genes as a result of genome-wide association studies with a recent meta-analysis listing 31 loci with confirmed evidence for association.1 What is remarkable is the degree of overlap of RA loci with loci identified in other autoimmune diseases including type 1 diabetes, systemic lupus erythematosus and coeliac disease, for example.2 ,3 Those autoimmune diseases are characterised by the presence of autoantibodies and differ from PsA in that respect. However, given the overlap of clinical features between RA and PsA, it might be expected that there would be some overlap in the genetic susceptibility.
The two major RA susceptibility genes are the HLA DRB1 and PTPN22 genes but previous investigations have largely reported no evidence for association with PsA.4,–,8 Few of the other loci have been investigated, to date. The aim of the current study was to investigate association of 41 suggestive and confirmed RA susceptibility loci with PsA in a large UK cohort.
Methods
Patient samples
A total of 1057 Genomic DNA samples collected from PsA patients of White European ancestry were available via the collaboration of three UK rheumatology centres and one centre in Ireland (885 UK and 172 Ireland), details of which have been described previously.9,–,11 PsA classification was defined as ‘an inflammatory arthritis associated with psoriasis, which is usually negative for rheumatoid factor’.12 This study was approved by the North West Multicentre Research Ethics Committee (MREC 99/8/84). All subjects provided informed consent.
Control samples
Single nucleotide polymorphism (SNP) genotype data were available for healthy controls from the 1958 British Birth Cohort and the UK Blood Service Collection. Both cohorts were genotyped on the Illumina Human1M-Duo and Affymetrix Genome-wide Human SNP Array 6.0 as part of the Wellcome Trust Case-Control Consortium 2 (WTCCC2) project (www.wtccc.org.uk). A total of 4000 genomic DNA samples were available for in-house genotyping of SNPs not represented on these arrays.
SNP selection
RA susceptibility SNPs were selected for genotyping if they were considered as confirmed associations or demonstrated suggestive evidence for association from a number of well-powered published reports.1 ,13,–,23
Genotyping
SNP genotyping of the PsA, 1958 birth cohort and Ireland control samples was performed using Sequenom's MassARRAY system (San Diego, California, USA) according to the manufacturers' specifications for the iPLEX chemistry using 10 ng of genomic DNA. Cluster plots for all SNPs were manually evaluated to confirm satisfactory performance. SNPs observed to have poor clustering characteristics were excluded from further analysis.
Statistical analysis
All quality control steps and statistical analyses were performed using the PLINK software package.24 Missing data rates for inclusion of both SNPs and samples were set at <10%. Test statistics for Hy–Weinberg equilibrium using an exact test, the Cochran-Armitage trend test and OR (including 95% CI) were calculated for the combined UK and Ireland dataset. To explicitly control for any bias introduced by population stratification, we analysed each population separately and combined the results via inverse-variance meta-analysis under the assumption of fixed effects. Allelic heterogeneity between the two groups was estimated using the Cochran Q and I2 statistics. A p value of <0.0015 was reged as statistically significant after applying a Bonferroni correction for the number of loci tested. Nominal associations were those at p <0.05.
Subphenotype analysis was performed within the PsA dataset based on, first, the age at onset of psoriasis (type I psoriasis has an onset ≤40 years of age while type II psoriasis is defined as an onset >40 years of age, n=354 and 540, respectively) and, second, seronegativity for rheumatoid factor (n=179) in an attempt to exclude those patients who may have PsV and coexisting RA. All subphenotype analyses were performed in UK samples only.
Results
SNP selection
A total of 56 SNPs mapping to 41 genomic regions previously reported as suggestive or confirmed susceptibility loci for RA were selected from published reports (see online supplementary table S1).
Genotyping
Eight of the selected SNPs failed inclusion during assay design and a further five SNPs were excluded due to unsatisfactory genotype clustering. Following the removal of samples and SNPs with high levels of missing data there were a maximum of 982 PsA cases, 2925 controls from the 1958 birth cohort (genotyped in-house), 371 Ireland controls and 5380 controls from the WTCCC2 data (see online supplementary table S2).
Statistical analysis
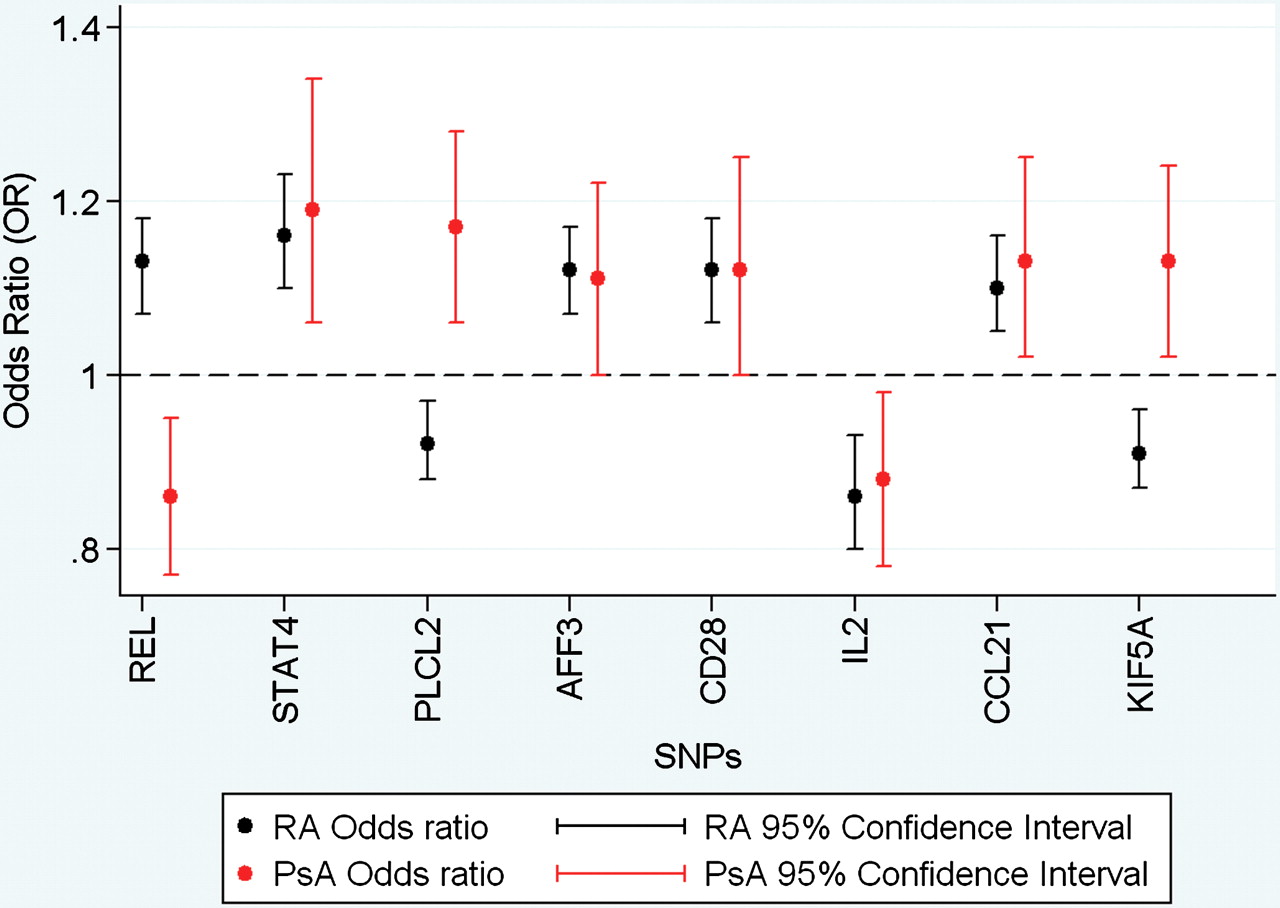
Investigation of the 43 successfully genotyped SNPs identified significant association (ptrend<0.0015) with one SNP, rs13017599 (REL), in the dataset as a whole (table 1) and when analysis was restricted to the UK dataset (ptrend=0.001, online supplementary table S2). The rs453211 (PLCL2) and rs10181656 (STAT4) SNPs were associated at nominal thresholds in the entire dataset (0.0016 and 0.003, respectively, table 1) and UK-only subgroups (ptrend=0.009, ptrend=0.02 respectively, online supplementary table S2). In addition, the analysis reveals nominal association (ptrend<0.05) to six SNPs: rs10865035 (AFF3), rs1980422 (CD28), rs2069778 (IL2), rs13192841 (TNFAIP3), rs2812378 (CCL21) and rs3184504 (KIF5A) in the combined UK and Ireland dataset but not when restricted to UK samples alone (table 1, online supplementary table S2). Interestingly, at three of the associated loci (REL, PLCL2 and STAT4) the direction of the effect was opposite to that reported in the RA studies (figure 1).
{kind=link}
OR plots for eight SNPs demonstrating evidence for association to PsA susceptibility, highlighting the opposing direction of effects for REL, PLCL2 and KIF5A. PsA, psoriatic arthritis; RA, rheumatoid arthritis, SNP, single nucleotide ploymorphism.
Association statistics for susceptibility to psoriatic arthritis for the 44 successfully genotype SNPs
Subphenotype analysis revealed a greater effect for the REL SNP, rs13017599, in the late onset psoriasis (type II) and the seronegative subgroups of PsA (see online supplementary table S3). Conversely, the association to the PLCL2 SNP, rs453211, was wholly restricted to the early onset psoriasis subgroup (type I) (see online supplementary table S3). While these associations are intriguing, their interpretation should be tempered by acknowledging the limited number of samples in these subgroups.
Discussion
We have undertaken a comprehensive analysis testing established RA susceptibility loci for association with PsA. We have found significant evidence for association with the REL locus and nominal evidence for association with seven other RA susceptibility SNPs. Interestingly, for three of the eight PsA-associated variants, the risk allele is opposite to that reported in RA.
Given the phenotypic similarities between RA and PsA, it was expected that some genetic overlap would be observed as the concept is well established for autoimmune diseases such as RA, systemic lupus erythematosus and type 1 diabetes.2 ,3 However, it could also be argued that inflammatory arthritis may be an outcome of different immune responses in joints caused by different triggers with different underlying genetic susceptibility. Indeed, enthesitis is thought to be the primary abnormality in PsA by many researchers, with synovitis being a secondary phenomenon, in contrast to RA where the synovitis takes primacy. The results of this genetic study support a mixed picture of genetic overlap between RA and PsA, with some RA loci showing association with the same allele, some with the opposite allele and some showing no association with PsA.
Association of different autoimmune diseases with opposite alleles of the same susceptibility variant has been reported previously. For example, the minor T allele of the PTPN22 rs2476601 SNP confers susceptibility to RA, type 1 diabetes and autoimmune thyroid disease while the major C allele confers susceptibility to Crohn's disease.25 Similarly, association at the REL locus SNP, rs13017599, has been reported previously in psoriasis but the opposite allele has also been associated with RA.26 ,27 These findings may suggest that RA clusters with the classical autoantibody associated autoimmune diseases, while PsA shares greater genetic similarity to psoriasis and seronegative diseases, such as Crohn's disease. It is possible that these SNPs may be markers for susceptibility to psoriasis and not specific to PsA. It would be of great interest to test these markers in a cohort of psoriasis patients screened to exclude samples with evidence of inflammatory arthritis.
The conclusions that can be drawn are necessarily limited by the limitations of the study design. Many of the RA susceptibility loci examined have modest effect sizes that the current study was underpowered to reliably detect online supplementary table S1. It may be that more RA susceptibility loci are associated with PsA than detected currently, therefore. For example, the study had only limited power to detect association at a number of the loci. This power is further reduced by using a Bonferroni corrected p value threshold and so there may be a number of false negative results. For this reason, we have reported loci showing nominal as well as significant evidence for association although replication is required for all these SNPs in additional datasets before they can be confidently labelled as PsA susceptibility loci. Ultimately, a full understanding of the extent of overlap between PsA and RA susceptibility loci will require comparison of well-powered genome-wide association studies in the two diseases.
A further limitation of the study design is the testing of only one or a small number of variants at each locus. It may be that different variants at the locus are more strongly associated with PsA; for example, different variants at the TNFAIP3 gene are associated with systemic lupus erythematosus, RA and psoriasis.18,28,–,31 Investigation of this possibility will require analysis of detailed fine mapping data of RA associated genomic loci in PsA samples.
The strongest evidence for association was with the REL locus where two SNPs showed association but only one remained significant at the corrected threshold (rs13017599). The REL locus, which encodes c-REL, a member of the NFκB inflammatory pathway, has been reported to be associated with type I psoriasis in a large genome-wide association study but with a different SNP, rs702873 (OR 1.12).27 There is strong, but not complete, correlation (r2=0.76 with rs13017599) between the two variants suggesting that the primary association is with psoriasis rather than PsA. The robust identification of PsA specific variants would require a collection of patients with uncomplicated psoriasis, where patients have been screened for the absence of inflammatory arthritis. Unfortunately, such a collection is not currently available to our research group.
Interestingly, the subphenotype analysis in the PsA samples suggests that the association may be even stronger in type II psoriasiscompared with the cohort as a whole (OR 1.47 vs 1.19, based on allele G as the risk allele) but this requires confirmation in other cohorts.
Association with PLCL2 has not been reported previously with psoriasis and it shows only borderline evidence for association in the current study when using the Bonferroni corrected p value. Furthermore, association of this locus with RA remains suggestive rather than confirmed at genome-wide significance thresholds and hence this result may represent a false positive finding. Nonetheless, it is of interest because the gene encodes a negative regulator of B cell receptor signalling, important in controlling immune responses and, again, the allele conferring risk to RA is protective for PsA.1 ,15
Of the other loci with nominal evidence for association, STAT4 has been reported to be associated with psoriasis previously in a Greek population.32 The reported SNP, rs7574865, is highly correlated with rs10181656 reported in this study (r2=1.00). Interestingly, the allele associated with RA susceptibility conferred protection to PsA. Association at the IL2/21 locus has been reported previously with a different SNP (rs13151961 r2=1.00) in a US PsA cohort33 and with the same SNP in a UK psoriasis cohort.34
An important point is that the majority of RA risk loci are identified using patients positive for anticyclic citrullinated peptide antibodies. Given the importance of seronegativity in the classification of PsA, it would be interesting to evaluate susceptibility risk loci identified in anticyclic citrullinated peptide antibody negative RA samples. However, to date there are no robustly confirmed susceptibility loci for seronegative RA
In summary, we report significant evidence for association of the REL locus with PsA and nominal evidence for association with eight other RA associated SNPs. For a significant minority of the loci, opposing alleles confer risk to PsA and RA suggesting that there are fundamental differences in the aetiological mechanisms underlying these two types of inflammatory arthritis.
Acknowledgments
The authors acknowledge the support of the NIHR Manchester Biomedical Research Centre and NIHR Leeds Musculoskeletal Biomedical Research Unit. John Bowes, Ian Bruce and Anne Barton are funded by Arthritis Research UK (arc grant; 17552). Edw Flynn is supported by the European Community's Sixth Framework Programme AutoCure funding. This study makes use of data generated by the Wellcome Trust Case-Control Consortium. A full list of the investigators who contributed to the generation of the data is available from www.wtccc.org.uk. Funding for the project was provided by the Wellcome Trust under aw 076113 and 085475.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Funding This work is supported by Arthritis Research UK.
Competing interests None.
Ethics approval North West Multicentre Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.