Article Text

Abstract
Objectives Axial spondyloarthritis (axSpA) is a complex disease with diverse manifestations, for which new treatment options are warranted. BE MOBILE 1 (non-radiographic (nr)-axSpA) and BE MOBILE 2 (radiographic axSpA (r-axSpA)) are double-blind, phase 3 trials designed to evaluate efficacy and safety of bimekizumab, a novel dual interleukin (IL)-17A and IL-17F inhibitor, across the axSpA spectrum.
Methods In parallel 52-week trials, patients with active disease were randomised 1:1 (nr-axSpA) or 2:1 (r-axSpA) to bimekizumab 160 mg every 4 weeks:placebo. From week 16, all patients received bimekizumab 160 mg every 4 weeks. Primary (Assessment of SpondyloArthritis international Society ≥40% improvement (ASAS40)) and secondary endpoints were assessed at week 16. Here, efficacy and treatment-emergent adverse events (TEAEs) are reported up to week 24.
Results 254 patients with nr-axSpA and 332 with r-axSpA were randomised. At week 16, primary (ASAS40, nr-axSpA: 47.7% bimekizumab vs 21.4% placebo; r-axSpA: 44.8% vs 22.5%; p<0.001) and all ranked secondary endpoints were met in both trials. ASAS40 responses were similar across TNFi-naïve and TNFi-inadequate responder patients. Improvements were observed in Ankylosing Spondylitis Disease Activity Score (ASDAS) states and objective measures of inflammation, including high-sensitivity C-reactive protein (hs-CRP) and MRI of the sacroiliac joints and spine. Most frequent TEAEs with bimekizumab (>3%) included nasopharyngitis, upper respiratory tract infection, pharyngitis, diarrhoea, headache and oral candidiasis. More fungal infections (all localised) were observed with bimekizumab vs placebo; no major adverse cardiovascular events (MACE) or active tuberculosis were reported. Incidence of uveitis and adjudicated inflammatory bowel disease was low.
Conclusions Dual inhibition of IL-17A and IL-17F with bimekizumab resulted in significant and rapid improvements in efficacy outcomes vs placebo and was well tolerated in patients with nr-axSpA and r-axSpA.
- Biological Therapy
- Inflammation
- Spondylitis, Ankylosing
- Cytokines
- Autoimmune Diseases
Data availability statement
Data are available on reasonable request. Underlying data from this manuscript may be requested by qualified researchers six months after product approval in the USA and/or Europe, or global development is discontinued, and 18 months after trial completion. Investigators may request access to anonymised individual patient-level data and redacted trial documents which may include: analysis-ready datasets, trial protocols, annotated case report forms, statistical analysis plans, dataset specifications, and clinical study reports. Prior to use of the data, proposals need to be approved by an independent review panel at www.Vivli.org and a signed data sharing agreement will need to be executed. All documents are available in English only, for a prespecified time, typically 12 months, on a password-protected portal.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
There remains a need for more treatment options with novel modes of action for axial spondyloarthritis (axSpA) as many patients fail treatment, do not achieve treatment targets or experience residual symptoms.
Bimekizumab, a monoclonal antibody which selectively inhibits interleukin (IL)-17F in addition to IL-17A, has demonstrated sustained efficacy and was well tolerated up to 3 years in patients with active ankylosing spondylitis (AS) in the phase 2b BE AGILE trial and its open-label extension.
WHAT THIS STUDY ADDS
These two parallel trials provide the first phase 3 data for the dual inhibition of IL-17A and IL-17F with bimekizumab, and cover the full spectrum of axSpA in patients with non-radiographic (nr)-axSpA (BE MOBILE 1) and r-axSpA (ie, AS; BE MOBILE 2).
Inhibition of IL-17F in addition to IL-17A with bimekizumab resulted in rapid, clinically relevant improvements in efficacy outcomes vs placebo starting after 1–2 weeks and was well tolerated.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Results from the BE MOBILE trials add to the growing evidence base supporting dual inhibition of IL-17A and IL-17F with bimekizumab as a novel therapeutic option for the treatment of axSpA.
Introduction
Axial spondyloarthritis (axSpA) is a chronic immune-mediated inflammatory disease mainly affecting the sacroiliac joints (SIJ) and spine, with additional peripheral and extramusculoskeletal manifestations.1–3 The axSpA spectrum encompasses patients with definitive structural damage to the SIJ visible on plain radiographs (radiographic (r-)axSpA, also known as ankylosing spondylitis4), and patients without definite radiographic sacroiliitis (non-radiographic (nr-)axSpA). Typically starting in patients’ mid-20s,5 axSpA has a significant and lasting impact on patient lives6–8 and a high disease burden.3 Many patients with axSpA fail treatment, do not achieve treatment targets or experience residual symptoms,9–11 demonstrating the need for more treatment options with novel modes of action.
Interleukin (IL)-17A and IL-17F are key mediators of inflammation.3 12 Signalling through the IL-17 receptor complex, IL-17A and IL-17F share >50% structural homology, and form homodimers and heterodimers (IL-17A/A; 17 A/F; 17 F/F) that can promote inflammation and bone formation/damage.13–15 Bimekizumab is a humanised monoclonal IgG1 antibody that selectively inhibits IL-17F in addition to IL-17A. Unlike IL-17A-specific inhibitors, bimekizumab enables neutralisation of IL-17A/A, IL-17A/F and IL-17F/F, which preclinical data and clinical trials comparing bimekizumab with IL-17A-specific inhibition in psoriasis suggest may lead to more effective suppression of inflammation.13 15–17
In the phase 2b BE AGILE trial in patients with active ankylosing spondylitis and its open-label extension, bimekizumab 160 mg every 4 weeks led to rapid disease control compared with placebo, with efficacy sustained for up to 3 years of treatment.18 19 Here, we present the first phase 3 efficacy and safety data for bimekizumab across the full axSpA spectrum from two parallel trials in patients with nr-axSpA (BE MOBILE 1) and r-axSpA (BE MOBILE 2).
Methods
Trial designs and oversight
These phase 3, multicentre, randomised, double-blind, placebo-controlled trials of bimekizumab were conducted in parallel, each at 83 sites across 14 countries in Asia, Eastern Europe, Western Europe and North America. Adults aged ≥18 years with active axSpA, defined as Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ≥4 and spinal pain (BASDAI item 2) ≥4, were screened for eligibility.
In BE MOBILE 1, patients had nr-axSpA as determined by clinical diagnosis and by fulfilling Assessment of SpondyloArthritis international Society (ASAS) classification criteria.20 Patients with nr-axSpA were also required to have objective inflammation at screening, specifically active sacroiliitis on MRI fulfilling the ASAS criteria (MRI+)21 and/or elevated C-reactive protein (CRP+) ≥6.0 mg/L. In BE MOBILE 2, patients had r-axSpA and fulfilled modified New York (mNY) criteria,22 including documented radiographic evidence of sacroiliitis (grade ≥2 bilateral or grade ≥3 unilateral); prior to enrollment, at the randomisation stage, fulfilment of the ASAS classification criteria was also checked and all patients in BE MOBILE 2 met both mNY and ASAS classification criteria.20 In both trials, prior failure of ≥2 non-steroidal anti-inflammatory drugs (NSAIDs), or history of intolerance or contraindication to NSAIDs was required. Patients were excluded if they had received >1 tumour necrosis factor inhibitor (TNFi), >2 additional biologic response modifiers (other than TNFis; including investigational biologics received in prior clinical trials), or any IL-17 response modifier. Patients who had previously received a TNFi must have been intolerant or experienced an inadequate response (IR) to previous treatment given at an approved dose for at least 12 weeks. Full exclusion criteria are provided in the online supplemental appendix.
Supplemental material
Most sites participated in both trials, allowing a cross-trial screening approach dependent on the presence or absence of definite sacroiliitis (according to mNY criteria)22 on plain radiographs. Radiographs were centrally read by two independent readers required to independently agree on the imaging evaluation, with an adjudicator in case of disagreement. Eligible patients without definite sacroiliitis were assigned to BE MOBILE 1; those with definite sacroiliitis to BE MOBILE 2.
Each trial included a 16-week placebo-controlled, double-blind treatment period (DBTP), followed by a 36-week active-treatment maintenance period (online supplemental figure S1). After screening, patients were randomised 1:1 (nr-axSpA; BE MOBILE 1) or 2:1 (r-axSpA; BE MOBILE 2) to receive bimekizumab 160 mg or placebo every 4 weeks to week 16, then bimekizumab 160 mg every 4 weeks from weeks 16 to 52. Randomisation was performed using interactive response technology, stratified by MRI/CRP classification and region in BE MOBILE 1, and by previous TNFi exposure and region in BE MOBILE 2 (online supplemental appendix). Bimekizumab and placebo were administered subcutaneously via a 1 mL prefilled syringe by unblinded trial personnel who were not involved in other trial aspects.

Key efficacy outcomes over time. Randomised set. aPrimary endpoint; bRanked secondary endpoint. Error bars show SE. All statistical tests were performed at a two-sided alpha level of 0.05. For binary endpoints, p values were calculated by logistic regression with treatment, MRI/CRP classification and region (BE MOBILE 1) or treatment, prior TNFi exposure and region (BE MOBILE 2) as factors. For continuous endpoints, p values were obtained by ANCOVA with treatment, MRI/CRP classification and region (BE MOBILE 1) or treatment, prior TNFi exposure and region (BE MOBILE 2) as fixed effects, and baseline values as covariates. ***p<0.001. ANCOVA, analysis of covariance; ASAS40, Assessment of Spondyloarthritis international Society 40% response; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; BKZ, bimekizumab; CfB, change from baseline; CRP, C-reactive protein; MI, multiple imputation; nr-axSpA, non-radiographic axial spondyloarthritis; NRI, non-responder imputation; PBO, placebo; Q4W, every 4 weeks; r-axSpA, radiographic axial spondyloarthritis; TNFi, tumour necrosis factor inhibitor.
Patient and public involvement
Patients with axSpA were consulted during development of the bimekizumab in axSpA clinical trial programme to understand treatment needs and recommend ways to facilitate trial participation while minimising burden of trial visits. Most efficacy endpoints were derived from existing patient-reported outcome measures which were originally developed with patient input to capture the experience of patients with axSpA. Trial participants were recruited by the trial sites and provided written consent to participate.
Endpoints
The primary efficacy endpoint in both trials was Assessment of SpondyloArthritis international Society ≥40% improvement (ASAS40) response at week 16.23 Ranked secondary efficacy endpoints were also assessed at week 16; to control family-wise type I error, primary and secondary ranked endpoints (online supplemental figure S2) were assessed in predefined sequences at α=0.05 (two-sided) until the first non-significant result. Other prespecified endpoints of key clinical importance included ASAS40 responses in TNFi-naïve (ranked secondary endpoint in BE MOBILE 2) and TNFi-IR patients, Ankylosing Spondylitis Disease Activity Score (ASDAS) states, change from baseline (CfB) in Maastricht Ankylosing Spondylitis Enthesitis Index (MASES) score (in patients with MASES>0 at baseline),24 complete resolution of enthesitis (MASES=0 in subset of patients with enthesitis (MASES>0) at baseline), CfB in swollen joint count (SJC) and tender joint count (TJC), and objective measures of inflammation including ratio to baseline in high-sensitivity CRP (hs-CRP), CfB in the Berlin modification of the spine ASspiMRI-a score (hereafter termed MRI Berlin spine score)25 and CfB in MRI Spondyloarthritis Research Consortium of Canada (SPARCC) SIJ inflammation score.26 BASDAI Q1 fatigue was not a prespecified endpoint but is reported here as part of the ASAS-OMERACT core domain set for axSpA.27 28 MRI endpoints are presented for the subset of patients in the MRI substudies at week 16, assessed by central reading.
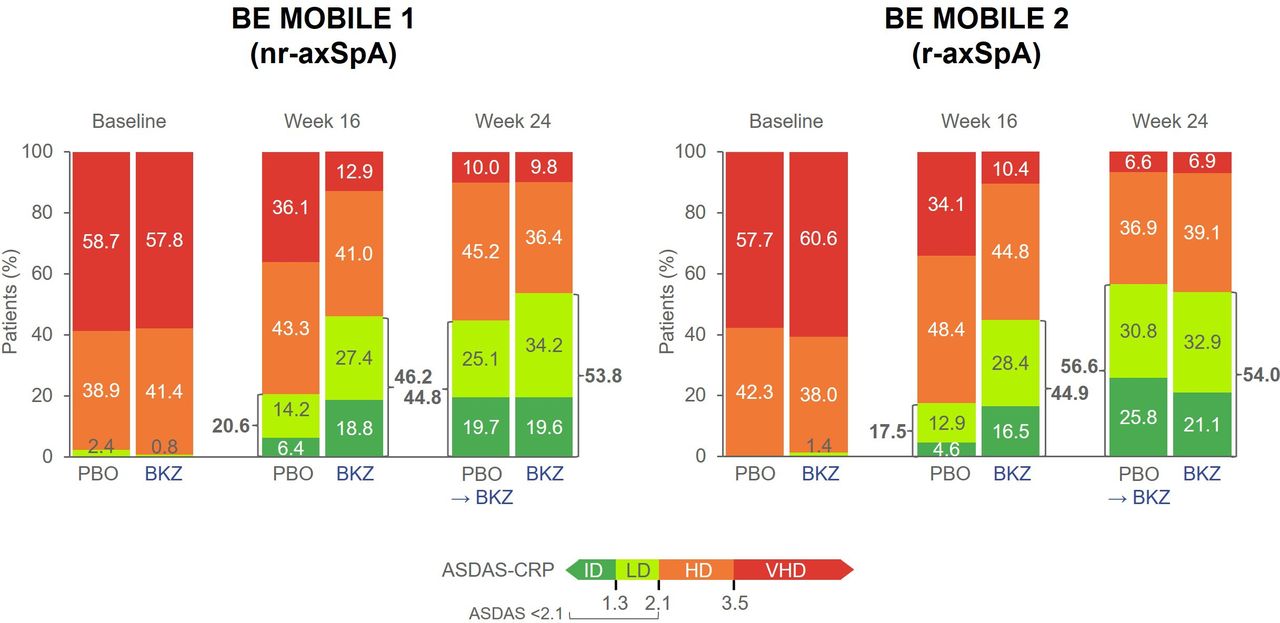
ASDAS disease states over time. Randomised set. Exploratory endpoint. Data reported are MI. VHD: ASDAS >3.5; HD: ASDAS ≥2.1 to ≤3.5; LD: ASDAS ≥1.3 to <2.1; ID: ASDAS <1.3. ASDAS, Ankylosing Spondylitis Disease Activity Score; BKZ, bimekizumab; HD, high disease; ID, inactive disease; LD, low disease; MI, multiple imputation; nr-axSpA, non-radiographic axial spondyloarthritis; PBO, placebo; Q4W, every 4 weeks; r-axSpA, radiographic axial spondyloarthritis; VHD, very high disease.
Incidence of treatment-emergent adverse events (TEAEs), treatment-emergent serious adverse events (SAEs) and TEAEs leading to withdrawal from the trial drug were all prespecified secondary endpoints. TEAEs, SAEs and prespecified safety topics of interest are defined in the online supplemental appendix. Adverse events were coded according to the Medical Dictionary for Regulatory Activities (MedDRA V.19.0).
Statistical analysis
In both trials, sample size calculations were based on testing of bimekizumab vs placebo for ASAS40 response at week 16. Efficacy results are reported at week 16 with summary data up to week 24 for each trial. All analyses were performed on the randomised set. Statistical analyses are detailed further in the online supplemental appendix.
For binary response endpoints (including the primary endpoint), missing data were treated as non-response. For continuous ranked endpoints reported at week 16, missing data to week 16 were imputed with reference-based multiple imputation, with the multiple imputation (MI) model based on placebo group data only. Missing data for non-ranked continuous endpoints and continuous ranked endpoints before and beyond week 16 were handled with MI using data from both bimekizumab and placebo groups. All between-group differences are adjusted risk differences from the logistic regression model for binary endpoints or mean differences vs placebo from the analysis of covariance (ANCOVA) model for continuous endpoints, with associated p values and 95% confidence intervals (CIs). The results for all endpoints that were not part of the statistical hierarchy are summarised with point estimates and are not controlled for multiplicity. For these non-ranked endpoints, MI was used for missing data in ASDAS states, hs-CRP, CfB in MASES score, CfB in SJC and TJC and BASDAI Q1 fatigue. Non-responder imputation (NRI) was used for missing data in ASAS40 responses in TNFi-naïve (non-ranked secondary endpoint in BE MOBILE 1) and TNFi-IR patients, complete resolution of enthesitis, and binary SJC and TJC measures (SJC=0 (in patients with SJC>0 at baseline) and TJC=0 (in patients with TJC>0 at baseline)). Observed case (OC) analyses were used for MRI Berlin spine and MRI SPARCC SIJ inflammation scores.
Safety for the DBTP (weeks 0–16) was summarised by treatment group for patients who had ≥1 dose of bimekizumab or placebo, respectively (safety set). Total adverse events from weeks 0–24 include adverse events occurring in patients who had ≥1 dose of bimekizumab during this period; TEAEs while on placebo are not included for weeks 0–24 data. Exposure-adjusted incidence rates per 100 patient-years (PY) of exposure are presented.
Results
Patient disposition and baseline characteristics
From 25 April 2019, 781 patients were screened and 254 were randomised in BE MOBILE 1 (bimekizumab: 128; placebo: 126). In BE MOBILE 2, 612 patients were screened and 332 were randomised (bimekizumab: 221; placebo: 111). The last week 24 visits were 1 December 2021 (BE MOBILE 1) and 25 October 2021 (BE MOBILE 2). Overall, 244 (96.1%) patients in BE MOBILE 1 and 322 (97.0%) in BE MOBILE 2 completed the DBTP to week 16; 240 (94.5%) and 313 (94.3%) patients completed treatment up to week 24, respectively (online supplemental figure S3). In both trials, patient demographics and baseline disease characteristics were similar across treatment groups and reflective of active axSpA (table 1). COVID-19 had minimal impact on trial conduct and results (online supplemental appendix).

{kind=link}
{kind=link}
{kind=link}
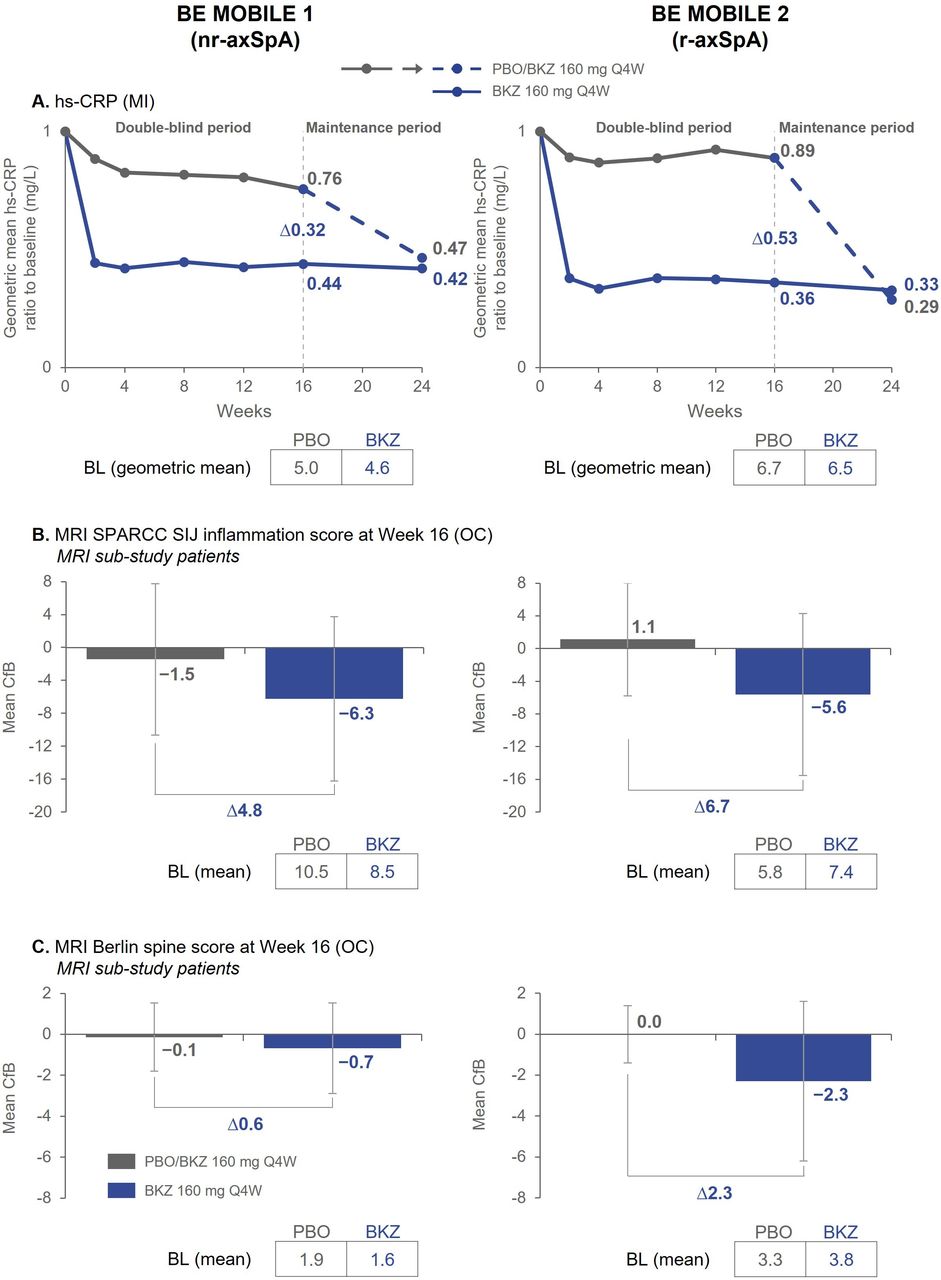
Objective signs of inflammation. Randomised set. Exploratory endpoints. Error bars show SD. (A) n=128 (BKZ) and n=126 (PBO) in BE MOBILE 1, n=221 (BKZ) and n=111 (PBO) in BE MOBILE 2; (B) At BL, n=79 (BKZ) and n=68 (PBO) in BE MOBILE 1, n=83 (BKZ) and n=45 (PBO) in BE MOBILE 2. At week 16, n=77 (BKZ), n=60 (PBO) in BE MOBILE 1, n=79 (BKZ) and n=43 (PBO) in BE MOBILE 2; (C) At BL, n=75 (BKZ) and n=65 (PBO) in BE MOBILE 1, n=82 (BKZ) and n=45 (PBO) in BE MOBILE 2. At week 16, n=73 (BKZ) and n=58 (PBO) in BE MOBILE 1, n=79 (BKZ) and n=43 (PBO) in BE MOBILE 2. MRI Berlin spine score ranges from 0 to 69; lower scores indicate less spinal inflammation and negative changes represent improvements. MRI SPARCC SIJ inflammation scores range from 0 to 72; lower scores indicate less SIJ inflammation and negative changes represent improvements. BKZ, bimekizumab; BL, baseline; CfB, change from baseline; hs-CRP, high-sensitivity C-reactive protein; MI, multiple imputation; nr-axSpA, non-radiographic axial spondyloarthritis; OC, observed case; PBO, placebo; Q4W, every 4 weeks; r-axSpA, radiographic axial spondyloarthritis; SIJ, sacroiliac joint; SPARCC, Spondyloarthritis Research Consortium of Canada.
Patient demographics and baseline characteristics
Efficacy at week 16
At the end of the DBTP, the primary endpoint was met in both studies (p<0.001; table 2; figure 1A), with 47.7% (61/128) vs 21.4% (27/126) patients with nr-axSpA and 44.8% (99/221) vs 22.5% (25/111) patients with r-axSpA achieving ASAS40 with bimekizumab and placebo, respectively. ASAS40 response rates in TNFi-naïve patients (secondary endpoint; ranked in BE MOBILE 2) were 45.7% (84/184) vs 23.4% (22/94) in patients with r-axSpA.
Efficacy outcomes at week 16 (primary and ranked secondary endpoints) and week 24
All ranked secondary endpoints were met in both trials with statistically significant improvements (table 2) observed with bimekizumab compared with placebo in ASAS responses (ASAS40 in TNFi-naïve patients (BE MOBILE 2), ASAS20, ASAS partial remission, ASAS 5/6), disease activity (BASDAI, ASDAS-MI), physical function (Bath Ankylosing Spondylitis Functional Index (BASFI), Short-Form 36-item Health Survey Physical Component Summary (SF-36 PCS)), pain (nocturnal spinal pain), quality of life (ASQoL) and spinal mobility (BASMI (BE MOBILE 2)).
Efficacy over time
Rapid separation (ie, within 1–2 weeks) was observed between bimekizumab and placebo in ASAS40 response rates in both trials, with differences between groups seen after one dose of treatment (figure 1A). In patients who received bimekizumab from baseline, the proportion of patients achieving ASAS40 response continued to increase to week 24 (nr-axSpA: 52.3%; r-axSpA: 53.8%), with improvements observed across all ASAS components comprising the primary endpoint (online supplemental table S1). For patients who switched from placebo to bimekizumab at week 16, week 24 ASAS40 responses reached similar levels to those seen in bimekizumab-randomised patients (nr-axSpA: 46.8%; r-axSpA 56.8%). Similar patterns of response were seen in all ranked secondary endpoints to week 24 in both trials (figure 1B,C; online supplemental figures S4,S5; table 2).
Exploratory endpoints
Almost all patients (≥97.6%) had high or very high disease activity (measured by ASDAS-CRP) at baseline. Improvements in the proportion of patients achieving ASDAS <2.1 (including low disease activity (≥1.3 to <2.1) and inactive disease (<1.3)) increased over time with bimekizumab treatment. At week 24, ASDAS<2.1 was achieved in over 50% of patients receiving bimekizumab from baseline (nr-axSpA: 53.8%; r-axSpA: 54.0%) and 44.8% and 56.6% of patients with nr-axSpA and r-axSpA, respectively, who switched from placebo to bimekizumab at week 16 (figure 2).
In both studies, rapid reduction of hs-CRP was observed with bimekizumab vs placebo as early as week 2 (first postbaseline measurement) and was maintained to week 24. Rapid hs-CRP reduction also occurred after switching from placebo to bimekizumab (figure 3A). At week 16, patients with nr-axSpA and r-axSpA in the MRI substudies demonstrated greater mean reductions from baseline in MRI SPARCC SIJ inflammation scores with bimekizumab vs placebo (nr-axSpA: –6.3 vs –1.5; r-axSpA: –5.6 vs +1.1, respectively; figure 3B). Mean MRI Berlin spine scores also decreased more from baseline with bimekizumab treatment vs placebo (nr-axSpA: –0.7 vs –0.1; r-axSpA: –2.3 vs 0.0, respectively; figure 3C).
As in patients with r-axSpA, ASAS40 response rates at week 16 in TNFi-naïve patients with nr-axSpA were greater with bimekizumab (46.6% (55/118)) than placebo (22.9% (25/109)). Similarly, more TNFi-IR patients achieved ASAS40 response with bimekizumab vs placebo at week 16 in both the nr-axSpA (60.0% (6/10) vs 11.8% (2/17)) and r-axSpA (40.5% (15/37) vs 17.6% (3/17)) populations.
Improvements in fatigue (measured by BASDAI Q1; online supplemental table S2), enthesitis (measured by MASES) and peripheral arthritis (measured by SJC and TJC) were also observed (online supplemental appendix).
Safety
Safety data are summarised in table 3. In the DBTP, 80/128 (62.5%) patients with nr-axSpA and 120/221 (54.3%) with r-axSpA receiving bimekizumab had ≥1 TEAE vs 71/126 (56.3%) and 48/111 (43.2%) receiving placebo, respectively. Through weeks 0–24, ≥1 TEAE was reported for 124/244 (50.8%) patients with nr-axSpA and 183/330 (55.5%) with r-axSpA who had received ≥1 dose of bimekizumab. TEAEs leading to trial drug discontinuation during weeks 0–24 occurred in 3/244 (1.2%) patients with nr-axSpA and 12/330 (3.6%) patients with r-axSpA (online supplemental table S3). No deaths occurred in either trial. SAEs are summarised in online supplemental table S4.
Safety overview for the double-blind treatment and overall periods
There were no serious infections in patients with nr-axSpA and 4/330 (1.2%) in bimekizumab-treated patients with r-axSpA during weeks 0–24; none led to discontinuation. Fungal infections were reported in 18/244 (7.4%) and 21/330 (6.4%) bimekizumab-treated patients with nr-axSpA and r-axSpA, respectively, during weeks 0–24 (table 3). All fungal infections were non-systemic and mucocutaneous, mild to moderate and none were classified as serious. The majority were Candida infections (nr-axSpA: 10/18; r-axSpA: 12/21), the most frequent preferred term being oral candidiasis (nr-axSpA: 7/18; r-axSpA: 10/21). Of patients with Candida infections, almost all were considered related to treatment (nr-axSpA: 9/10; r-axSpA: 11/12), however, none of those with nr-axSpA and 2 (1 oral and 1 oesophageal candidiasis) with r-axSpA discontinued treatment. Among bimekizumab-treated patients, there were few infections classified as opportunistic in either trial during weeks 0–24 (nr-axSpA: 3/244 (1.2%); r-axSpA: 2/330 (0.6%)); all were localised fungal infections, with no cases of active tuberculosis.
Among prespecified safety topics of interest during weeks 0–24, no anaphylactic reactions, serious hypersensitivity reactions or adjudicated major adverse cardiovascular events occurred. There was one malignancy (clear cell renal cell carcinoma) in a bimekizumab-treated patient with nr-axSpA, and 1 case of suicidal ideation in a bimekizumab-treated patient with r-axSpA was adjudicated as suicidal ideation and behaviour. Non-serious neutropenia TEAEs with bimekizumab occurred in 2 patients in each trial (nr-axSpA: 0.8%; r-axSpA: 0.6%); none of these patients developed a concurrent infection or discontinued treatment. Hepatic events occurred in 11/244 (4.5%) and 16/330 (4.8%) patients with nr-axSpA and r-axSpA, respectively. The majority of hepatic events were non-serious liver function elevations, and none led to treatment discontinuation. Those that were markedly abnormal were generally associated with factors other than trial treatment.
During weeks 0–24, no bimekizumab-treated patients with nr-axSpA had a TEAE adjudicated to be probable or definite inflammatory bowel disease (IBD). In bimekizumab-treated patients with r-axSpA, 1 patient (0.3%) had moderate Crohn’s disease leading to discontinuation from the trial and 1 patient (0.3%) had severe ulcerative colitis leading to discontinuation of trial drug (both new-onset). During the DBTP, uveitis events in patients with nr-axSpA were lower with bimekizumab than placebo (2 (1.6%; 1 new-onset) vs 6 (4.8%; 2 new-onset) patients, respectively), and only observed in the placebo group (5 (4.5%; none new-onset) patients) in patients with r-axSpA. During weeks 0–24, incidence of uveitis was low in bimekizumab-treated patients (nr-axSpA: 2 (0.8%; 2.6/100 PY); r-axSpA: 2 (0.6%; 1.7/100 PY)). Discontinuation of treatment due to uveitis occurred in only 2 placebo-treated patients with nr-axSpA; there were no serious uveitis events in bimekizumab-treated patients.
In each trial, there was one confirmed, non-serious COVID-19 infection during bimekizumab treatment in weeks 0–24 (further information on COVID-19 infections and potential impacts of the pandemic on the trials available in the online supplemental appendix).
Discussion
In these phase 3 trials, dual inhibition of IL-17A and IL-17F with subcutaneous bimekizumab 160 mg every 4 weeks led to significant improvements in ASAS responses, disease activity, physical function, pain, quality of life and spinal mobility compared with placebo in patients with nr-axSpA and r-axSpA. Consistent results across these parallel trials highlight the efficacy of bimekizumab across the full axSpA spectrum. They add to a growing evidence base demonstrating the efficacy of bimekizumab in spondyloarthritis, including the BE OPTIMAL and BE COMPLETE phase 3 trials in patients with psoriatic arthritis.29 30
The BE MOBILE trials also provide evidence to suggest that bimekizumab is efficacious in patients with axSpA regardless of prior TNFi exposure. Despite limited patient numbers, efficacy assessed by ASAS40 response was similar across TNFi-naïve and TNFi-IR patients, consistent with findings of comparable efficacy from recent phase 3 studies of bimekizumab in biologic-naïve and TNFi-IR patients with psoriatic arthritis.29 30 Biomarker analysis in psoriatic arthritis has demonstrated an IL-17F signal in biologic-IR patients,31 providing a possible explanation for the similar efficacy in this subgroup and further highlighting the potential of dual IL-17A/IL-17F inhibition with bimekizumab.
Bimekizumab was associated with improvements in exploratory analyses of objective disease measures, consistent with the results for the primary and secondary analyses of patient-reported outcomes. Substantial improvements were observed in objective signs of inflammation (hs-CRP and MRI scores) with bimekizumab vs placebo, which are predictors for progression of structural damage in axSpA.1–3 Additionally, the proportion of patients achieving ASDAS low disease activity or inactive disease (ASDAS<2.1; a key treatment target in axSpA)2 3 increased from <2% to >50% with 24 weeks of bimekizumab treatment, demonstrating that bimekizumab can lead to a large proportion of patients meeting clinically relevant treatment targets.
Bimekizumab was generally well tolerated in patients with nr-axSpA and r-axSpA; no new safety signals were observed. Consistent with other phase 3 trials of bimekizumab and the known role of IL-17 in host defence against Candida at the mucosal barrier,17 32 33 oral candidiasis was more common with bimekizumab than placebo. Most fungal infections with bimekizumab were mild to moderate, and none were systemic or led to treatment discontinuation. Incidence of uveitis, the most common extramusculoskeletal manifestation of axSpA,3 was lower with bimekizumab than placebo in the DBTP and remained low to week 24. Although overall exposure was limited and further investigation is needed, this observation aligns with prior observations.18 19 Incidence of IBD, another key extramusculoskeletal manifestation,1 was also low with bimekizumab, suggesting inhibition of IL-17F in addition to IL-17A does not exacerbate IBD compared with inhibition of IL-17A alone.2 32–34
Strengths of these trials include coverage of the full spectrum of axSpA (including the first trial of bimekizumab in nr-axSpA) with a parallel, cross-trial screening approach and central reading of MRIs and radiographs. A limitation of these analyses is the 24-week duration being too brief to assess longer-term efficacy and safety of bimekizumab. Results beyond week 16 should be interpreted with caution due to patients’ awareness of receiving active treatment after week 16, as should exploratory endpoints, which were not controlled for multiplicity. Direct comparisons between bimekizumab and other IL-17 inhibitors cannot be made using these placebo-controlled trial data from BE MOBILE 1 and 2; head-to-head trials would be required for such comparisons.
In summary, these first phase 3 trials evaluating the efficacy and safety of dual inhibition of IL-17F in addition to IL-17A with bimekizumab, across the full spectrum of axSpA, clearly demonstrate that treatment with bimekizumab resulted in rapid, clinically relevant improvements in disease manifestations, compared with placebo, and was well tolerated. Bimekizumab may therefore offer patients with axSpA an effective treatment option with a novel mode of action.
Data availability statement
Data are available on reasonable request. Underlying data from this manuscript may be requested by qualified researchers six months after product approval in the USA and/or Europe, or global development is discontinued, and 18 months after trial completion. Investigators may request access to anonymised individual patient-level data and redacted trial documents which may include: analysis-ready datasets, trial protocols, annotated case report forms, statistical analysis plans, dataset specifications, and clinical study reports. Prior to use of the data, proposals need to be approved by an independent review panel at www.Vivli.org and a signed data sharing agreement will need to be executed. All documents are available in English only, for a prespecified time, typically 12 months, on a password-protected portal.
Ethics statements
Patient consent for publication
Ethics approval
The BE MOBILE 1 and BE MOBILE 2 trials were conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation Guidance for Good Clinical Practice. Ethical approvals were obtained from the relevant institutional review boards at participating sites. All patients provided written informed consent in accordance with local requirements. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors thank the patients and their caregivers in addition to all the investigators and their teams who contributed to these studies. The authors also acknowledge Natasha de Peyrecave, DPhil, UCB Pharma, Brussels, Belgium for substantial contributions to this publication, Celia Menckeberg, PhD, UCB Pharma, Breda, The Netherlands for publication coordination and editorial assistance, and Evelyn Turner, BSc, Costello Medical, Cambridge, UK for medical writing and editorial assistance based on the authors’ input and direction. These trials were funded by UCB Pharma.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
DvdH and AD are joint first authors.
Handling editor Josef S Smolen
Contributors Substantial contributions to study conception and design: DvdH, AD, XB, MAB, HD, MD, DE, AME, CF, KG, LSG, NH, MM, WPM, AM, UM, MO, DP, MR, JSS, TT, FVdB, TV and HX; substantial contributions to analysis and interpretation of the data: DvdH, AD, XB, MAB, HD, MD, DE, AME, CF, KG, LSG, NH, MM, WPM, AM, UM, MO, DP, MR, JSS, TT, FVdB, TV and HX; drafting the article or revising it critically for important intellectual content: DvdH, AD, XB, MAB, HD, MD, DE, AME, CF, KG, LSG, NH, MM, WPM, AM, UM, MO, DP, MR, JSS, TT, FVdB, TV and HX; final approval of the version of the article to be published: DvdH, AD, XB, MAB, HD, MD, DE, AME, CF, KG, LSG, NH, MM, WPM, AM, UM, MO, DP, MR, JSS, TT, FVdB, TV and HX; manuscript guarantor: DvdH.
Funding This article was based on the original trials BE MOBILE 1 (NCT03928704) and BE MOBILE 2 (NCT03928743) which were sponsored by UCB Pharma. Support for third party medical writing assistance for this article, provided by Evelyn Turner, BSc, Costello Medical, Cambridge, UK was funded by UCB Pharma in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Competing interests DvdH: Consulting fees from AbbVie, Bayer, BMS, Cyxone, Eisai, Galapagos, Gilead, GSK, Janssen, Lilly, Novartis, Pfizer, UCB Pharma, and is the director of Imaging Rheumatology BV; AD: Speaker for Janssen, Novartis, and Pfizer; consultant of AbbVie, Amgen, Aurinia, BMS, Celgene, Eli Lilly, GSK, Janssen, MoonLake, Novartis, Pfizer, and UCB Pharma; grant/research support from AbbVie, Eli Lilly, GSK, Novartis, Pfizer, and UCB Pharma; XB: Speaker for AbbVie, BMS, Chugai, Eli Lilly, Galapagos, Gilead, MSD, Novartis, Pfizer, and UCB Pharma; paid instructor for AbbVie, BMS, Chugai, Eli Lilly, Galapagos, Gilead, MSD, Novartis, Pfizer, and UCB Pharma; consultant for AbbVie, BMS, Chugai, Eli Lilly, Galapagos, Gilead, MSD, Novartis, Pfizer, and UCB Pharma; MAB: Grant/research support from UCB; Consultant for Clementia, Grey Wolf Therapeutics, Incyte, Ipsen, Pfizer, Regeneron, and Xinthera; Speaker for Novartis; HD: Speaker for BMS, Chugai, Eli Lilly, GSK, MSD, Novartis, Pfizer, UCB Pharma; MD: Consultant for AbbVie, Eli Lilly, Novartis, Merck, Pfizer, and UCB Pharma; Grant/research support from: AbbVie, Eli Lilly, Novartis, Pfizer, and UCB Pharma; DE: Consultancy and speaker fees from AbbVie, Eli Lilly, Galapagos, Novartis and UCB Pharma; KG: Consultant of AbbVie, Eli Lilly, Novartis, and UCB Pharma; grant/research support from AbbVie, Gilead, Eli Lilly, Novartis, and UCB Pharma; speakers bureau from AbbVie, Eli Lilly, Novartis, UCB Pharma; LSG: Consulting fees from AbbVie, Eli Lilly, Gilead, Janssen, MoonLake, Novartis, Pfizer, and UCB Pharma; grant/research support from Novartis, Pfizer and UCB Pharma; NH: Consulting fees from AbbVie, Eli Lilly, Janssen, Novartis and UCB Pharma; MM: Consultancy fees from AbbVie, BMS, Eli Lilly, Novartis, Pfizer and UCB Pharma, and research grants from AbbVie, BMS and UCB Pharma; WPM: Honoraria/consulting fees from AbbVie, Boehringer-Ingelheim, Celgene, Eli Lilly, Galapagos, Janssen, Novartis, Pfizer and UCB Pharma; research grants from AbbVie, Pfizer; educational grants from AbbVie, Janssen, Novartis and Pfizer; Chief Medical Officer for CARE Arthritis; DP: Speaker for AbbVie, BMS, Eli Lilly, MSD, Novartis, Pfizer, and UCB Pharma; Consultant for AbbVie, Biocad, Eli Lilly, Gilead, GSK, MSD, Novartis, Pfizer, Samsung Bioepis, and UCB Pharma; Grant/research support from: AbbVie, MSD, Novartis, and Pfizer; MR: Speakers bureau from AbbVie, BMS, Boehringer Ingelheim, Chugai, Eli Lilly, Janssen, Novartis, Pfizer, UCB Pharma; consultant of AbbVie, Eli Lilly, Novartis, UCB Pharma; TT: Consultancy fees: AbbVie, Eli Lilly, Gilead, Novartis, and Pfizer; Speaker fees: AbbVie, Astellas, BMS, Eisai, Eli Lilly, Janssen, Kyowa Kirin, Mitsubishi-Tanabe, Novartis, and Pfizer; FVdB: Consultancy fees from AbbVie, Amgen, Eli Lilly, Galapagos, Janssen, Merck, Novartis, Pfizer and UCB Pharma; Speakers bureau fees from AbbVie, BMS, Celgene, Janssen, Merck, Novartis, Pfizer and UCB Pharma; HX: Speaker for AbbVie, Janssen, Novartis, Pfizer, and UCB Pharma; Consultant for AbbVie, Beigene, BioMap, IASO, Pfizer, and UCB Pharma; Clinical investigator for Peking-Tsinghua Center for Life Sciences; AM, UM, MO, CF, TV, AME, JSS: Employees of UCB Pharma.
Patient and public involvement Patients and/or the public were involved in the design, or conduct, or reporting, or dissemination plans of this research. Refer to the Methods section for further details.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.