Article Text

Abstract
Objectives To elucidate the mechanism of action of baricitinib, a Janus kinase (JAK) 1/2 inhibitor, and describe immunological pathways related to disease activity in adults with systemic lupus erythematosus (SLE) receiving standard background therapy in a phase II trial.
Methods Patients with SLE were treated with baricitinib 2 mg or 4 mg in a phase II randomised, placebo-controlled study. Sera from 239 patients (baricitinib 2 mg: n=88; baricitinib 4 mg: n=82; placebo: n=69) and 49 healthy controls (HCs) were collected at baseline and week 12 and analysed using a proximity extension assay (Target 96 Inflammation Panel (Olink)). Interferon (IFN) scores were determined using an mRNA panel. Analytes were compared in patients with SLE versus HCs and in changes from baseline at week 12 between baricitinib 2 mg, 4 mg and placebo groups using a restricted maximum likelihood-based mixed models for repeated measures. Spearman correlations were computed for analytes and clinical measurements.
Results At baseline, SLE sera had strong cytokine dysregulation relative to HC sera. C-C motif chemokine ligand (CCL) 19, C-X-C motif chemokine ligand (CXCL) 10, tumour necrosis factor alpha (TNF-α), TNF receptor superfamily member (TNFRSF)9/CD137, PD-L1, IL-6 and IL-12β were significantly reduced in patients treated with baricitinib 4 mg versus placebo at week 12. Inflammatory biomarkers indicated correlations/associations with type I IFN (CCL19, CXCL10, TNF-α and PD-L1), anti-double stranded DNA (dsDNA) (TNF-α, CXCL10) and Systemic Lupus Erythematosus Disease Activity Index-2000, tender and swollen joint count and worst joint pain (CCL19, IL-6 and TNFRSF9/CD137).
Conclusion These results suggest that baricitinib 4 mg downregulated key cytokines that are upregulated in patients with SLE and may play a role in a multitargeted mechanism beyond the IFN signature although clinical relevance remains to be further delineated.
Trial registration number NCT02708095.
- cytokines
- systemic lupus erythematosus
- inflammation
Data availability statement
Lilly provides access to all individual participant data collected during the trial, after anonymisation, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the US and EU and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
What is already known on this topic
Systemic lupus erythematosus (SLE) is made complex by various immunological abnormalities contributing to SLE pathogenesis.
In a phase II study of baricitinib in patients with SLE, daily oral baricitinib 4 mg in conjunction with current standard of care (SOC) was superior to placebo plus SOC in improving SLE disease activity at week 24.
What this study adds
Insights into the mechanism of action of baricitinib in SLE.
How this study might affect research, practice and or/policy
The analysis presented here advances the understanding of how baricitinib may act on clinically relevant pathways in patients with SLE.
These results also serve to elucidate potential new biological targets that may impact SLE pathogenesis.
Introduction
The clinical complexity of patients with systemic lupus erythematosus (SLE) is a reflection of various immunological abnormalities contributing to SLE pathogenesis, including dysregulation of both the innate and adaptive immune responses, leading to the breakdown of tolerance, production of autoantibodies, deposition of immune complexes in tissues, leading to the activation of complement and the accumulation of neutrophils, monocytes and self-reactive T and B-lymphocytes.1–3 Research into the pathogenesis of SLE offers a nexus of gene expression, cell signalling and cellular responses that can present with different degrees of dysregulation among patients with SLE. Key cytokines in SLE comprise, among others, type I interferon (IFN),4 type II IFN, IL-6, IL-12/23, IL-17 and B lymphocyte stimulator (BAFF/BlyS)5 representing the clinical and molecular heterogeneity of SLE. Abnormalities include alterations in the expression of IFN inducible chemokines, such as C-X-C motif chemokine ligand (CXCL) 10 and C-C motif chemokine ligand (CCL) 19 (which have been shown to correlate with disease activity),6 alterations in B cell receptor signalling and alterations in the expression of cytokines related to leucocyte, neutrophil and macrophage trafficking, such as IL-6 and others.3 7–9
In a phase II study of baricitinib in patients with SLE, daily oral baricitinib 4 mg in conjunction with current standard of care (SOC) was superior to placebo plus SOC in improving SLE disease activity at week 24.10 Microarray analysis on serum samples in this study cohort found that treatment with baricitinib 4 mg significantly reduced the RNA expression of a network of genes functionally interconnected in SLE (signal transducer and activator of transcription (STAT)1, STAT2 and STAT4-target genes and multiple IFN responsive genes). Furthermore, baricitinib downregulated cytokine signalling associated with SLE pathogenesis and the Janus kinase (JAK)/STAT pathway, such as IL-6 and IL-12.11 While recent phase III trials have shown inconclusive results for the efficacy of baricitinib to treat moderate to severe SLE adult patients, the objective of this study was to evaluate the pharmacodynamic effect of baricitinib on a broad and highly sensitive array of serum cytokines in patients with SLE and to characterise immune pathways involved in the mechanism of action of baricitinib.
Methods
Study design
Patient samples were obtained from the double-blind, multicentre, randomised, placebo-controlled, 24-week phase II clinical trial, I4V-MC-JAHH.10 Eligible patients were aged 18 years or older and had a diagnosis of SLE. At baseline, patients were required to have a positive antinuclear antibody, or a positive anti-dsDNA, arthritis or rash as defined by Systemic Lupus Erythematosus Disease Activity Index-2000 (SLEDAI-2K) and a clinical SLEDAI-2K score of ≥4. Study drug was added to existing stable background SOC therapy, which could include corticosteroids up to 20 mg/day of prednisone or equivalent, a single antimalarial, a single immunosuppressant and/or non-steroidal anti-inflammatory drugs. Tapering of corticosteroids was permitted from baseline to week 16. Active central nervous system SLE or active severe SLE nephritis were not permitted.
This study was done in accordance with the ethical principles of the Declaration of Helsinki and Good Clinical Practise guidelines. All investigation sites received approval from the appropriate authorised institutional review board or ethics committee. All patients provided written consent before the study-related procedures were carried out.
Randomisation and masking
Patients were allocated (1:1:1) using a computer-generated random sequence to baricitinib 2 mg, baricitinib 4 mg or placebo plus SOC. Patients were stratified according to disease activity (SLEDAI-2K score <10 or ≥10), anti-dsDNA status (positive or negative) and region (USA, Europe, Asia or rest of the world). Investigators and patients were masked to allocation.
Serum cytokine quantification (proximity extension analysis)
Serum samples from 239 patients (baricitinib 2 mg: n=88; baricitinib 4 mg: n=82; placebo: n=69) were analysed with the Olink Inflammation I (95302) multiplex proximity extension assay (PEA) technology (Uppsala, Sweden) according to manufacturer’s specifications. The levels of analyte-specific DNA amplicons for 92 soluble analytes were quantified for each patient on the Fluidigm Biomark HD (San Francisco, California) at baseline and week 12. Serum samples from 49 age/sex-matched healthy controls (HCs) were included for comparisons to baseline SLE samples.
Statistical analyses
Comparisons between patients with SLE and HCs as well as those treated either with baricitinib 2 mg, baricitinib 4 mg or placebo plus SOC were made using a mixed-effect repeated measure model with an unstructured variance–covariance matrix and log2 transformed relative protein expression as the response. The lme4 function in R V.4.0.3 with fixed effect, covariates of sex, batch and corticosteroid use at baseline were used to fit the model.
A total of four different contrasts were tested
Patients with SLE compared with HCs at baseline.
Changes from baseline to week 12 compared between baricitinib 4 mg versus placebo plus SOC.
Changes from baseline to week 12 compared between baricitinib 2 mg versus placebo plus SOC.
Changes from baseline to week 12 compared between baricitinib 4 mg treatment versus baricitinib 2 mg treatment.
For within-protein multiplicity adjustment, the glht function12 was applied to all comparisons. The threshold of adjusted p value generated from within-protein multiplicity control to identify statistically significant proteins was 0.0237. For between-protein multiplicity adjustment, the q value was calculated with the smallest within-protein p value using a false discovery rate threshold set to 0.05. Spearman correlation was applied to key continuous clinical outcomes and protein expression levels with Benjamini-Hochberg multiple comparison adjustment to report the correlation values and adjusted p values. We used 0.05 as the cut-off value to determine the statistical significance of correlation coefficients.
Clinical correlations
IFN signature
Score was assessed previously using a validated mRNA panel.13
Anti-dsDNA serum levels
Serum samples were analysed for changes from baseline over time for anti-dsDNA antibodies using INOVA QUANTA Lite SC ELISA (INOVA Diagnostics, San Diego, California).10
SLEDAI-2K
The SLEDAI-2K14 is a validated global disease activity instrument that focuses on high-impact disease manifestations across nine organ systems. It includes 24 clinical and laboratory variables with manifestations graded by the affected organ system.
Worst joint pain
Worst joint pain was measured using the Brief Pain Inventory (short form) (BPI-sf)-modified worst joint pain item, which is a self-administered question developed for the rapid assessment of pain intensity. Worst joint pain item asks patients to rate their pain at its worst over the past 7 days.15
28 swollen and tender joint count
The 28 joints examined and assessed as tender or not tender for tender joint count and as swollen or not swollen for swollen joint count include 14 joints on each side of the patient’s body.16
Patient and public involvement statement
Patients were not involved in the study design.
Results
Analyte abnormalities in patients with SLE at baseline characterise cytokine dysregulation
Cytokine levels were analysed at baseline in patients with SLE and compared with HCs. Of the 92 detectable analytes measured, 17 were significantly upregulated (table 1) and 9 were downregulated in patients with SLE (online supplemental table S1). Of note, several chemokines such as CCL19, CXCL10, CXCL9, CCL2 and CCL20 and proinflammatory cytokines, such as IL-6, IL-12, IL-17A, were increased in patients with SLE versus HCs that, in addition to the increased PD-L1 and IL-10, indicate abnormalities of chronic SLE immunity.
Supplemental material
Upregulated analytes in patients with SLE versus HCs at baseline
Baricitinib modulates disturbances of cytokine networks in SLE
Treatment with baricitinib 4 mg significantly reduced the serum expression levels of 7 of the 17 initially increased analytes relative to placebo plus SOC in patients with SLE at 12 weeks (table 2) among others (online supplemental table S2).
Analytes upregulated in patients with SLE and downregulated by treatment with baricitinib 4 mg relative to placebo plus SOC at week 12
Baricitinib 4 mg treatment specifically and significantly downregulated serum cytokines that mediate lymphocyte and monocyte/macrophage recruitment such as chemokine (C-C motif) ligand 19 (CCL19), IFN-γ-induced proteins such as CXCL10, tumour necrosis factor (TNF) receptor superfamily member 9 (TNFRSF9) and TNF alpha (TNF-α) as well as IL-12β and IL-6 expression levels compared with placebo plus SOC at 12 weeks (table 2). Beyond direction of immune cells towards inflammatory sites, these molecules are relevant for B–T lymphocyte interactions (ie, PD-L1) confirming previous findings using multiplex cytokine panel quantitative (Quanterix) assays.11
Less pronounced decreases in similar cytokines were observed with baricitinib 4 mg versus 2 mg (online supplemental table S3). Of note, three analytes not typically associated with IFN signalling, TRANCE/CD254, TNFRSF9 and TNF-α, were reduced in the baricitinib 4 mg treatment group versus 2 mg treatment group.
On the other hand, only three analytes increased significantly (NT-3, SCF and CXCL5) (online supplemental table S2) under treatment with baricitinib 4 mg versus placebo plus SOC in patients with SLE, but not between the two baricitinib treatment groups (online supplemental table S3).
These results suggest that treatment with baricitinib 4 mg might mediate changes within the inflammatory JAK/STAT cytokine network beyond the IFN signature, considered a key molecular signature in SLE.
Clinical correlates and cytokine changes with baricitinib treatment in SLE
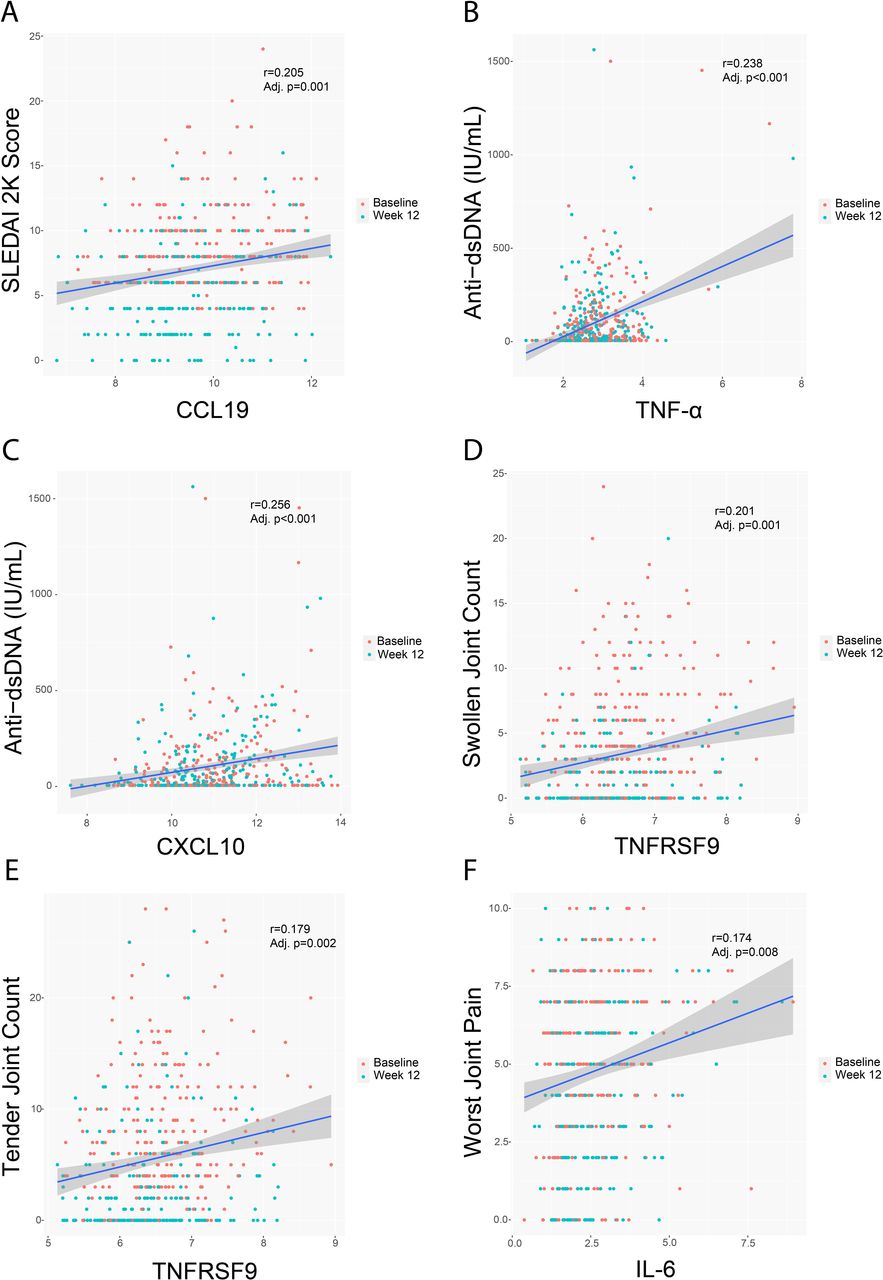
Certain cytokines downregulated by treatment with baricitinib 4 mg correlated with the IFN signature. The most representative ones were CCL19, CXCL10, TNF-α and soluble PD-L1/CD274 (figure 1). Correlation analysis identified a relationship between the observed cytokine/chemokine changes and clinical and serologic measures of SLE activity, including anti-dsDNA production (figure 2). Interestingly, in addition to positive correlations with the IFN signature, there was a significant positive correlation of CCL19 with SLEDAI-2K (figure 2). Significant positive correlations were also found between TNFRSF9 levels and swollen and tender joint counts and between IL-6 levels and worst joint pain (figure 2).

Correlation between key analytes and the IFN signature. Line of regression and confidence intervals are shown for CCL19 (A), CXCL10 (B), TNF-α (C), and PD-L1 (D) and the IFN signature. CCL19, C-C motif chemokine ligand 19; CXCL10, C-X-C motif chemokine ligand 10; IFN, interferon; PD-L1, PDCD1 ligand 1; TNF-α, tumour necrosis factor alpha.

{kind=link}
{kind=link}
Correlation between key analytes and clinical measurements at baseline and week 12. Line of regression and CIs are shown for CCL19 and SLEDAI-2K (A), TNF-α (B) and CXCL10 (C) and anti-dsDNA expression levels, TNFRSF9 and swollen (D) and tender (E) joint count, and between IL-6 and worst joint pain (F). CCL19, C-C motif chemokine ligand 19; CXCL10, C-X-C motif chemokine ligand 10; TNF-α, tumour necrosis factor alpha; IL-6, interleukin-6; TNFRSF9, TNF receptor superfamily member 9.
These data indicate that changes in cytokine expression in patients with SLE treated with baricitinib might be relevant to clinical outcome measures. Furthermore, although weak, the positive correlations seen between key cytokines and SLEDAI-2K, joint parameters and anti-dsDNA production suggest that the underlying mechanisms of cytokine modulation can exert an effect on joint pathology in patients with SLE as well as inhibit the B cell activation that results in antibody production. Further clinical evidence will be needed to confirm these observations.
Discussion
The goal of this study was to further understand SLE immunopathogenesis and elucidate how baricitinib might act by identifying key cytokines significantly downregulated after treatment and their potential correlations with clinical outcomes. First, we wanted to investigate any functional regulation of cytokines by baricitinib in patients with SLE, particularly any relating to previously identified gene associated changes11 and, second, we wanted to build on the original analysis using a multitargeted inflammatory panel with sensitivity to investigate a wider range of potential therapeutic targets of baricitinib. Such PEAs have been previously used to identify, at the protein level and as such more close to functional consequences than mRNA transcripts, key analytes deregulated and their correlation with clinical outcomes and organ damage in SLE.17–20
Specifically, for our study, as previously shown, baricitinib treatment reduced the mRNA expression of functionally interconnected genes involved in SLE including STAT1, STAT2 and STAT4-targeting genes as well as multiple IFN responsive genes. Baricitinib also reduced serum levels of two key cytokines implicated in SLE pathogenesis, IL-6 and IL-12β.11 Expanding on these preliminary findings, in the new analyses presented here, we detected seven analytes that were significantly elevated at baseline in patients with SLE compared with HCs and significantly reduced at week 12 after treatment with baricitinib 4 mg, including the previously identified IL-6 and IL-12β as well as other cytokines not typically associated with JAK-STAT signalling such as CXCL10, CCL19, TNFRSF9, TNF-α and PD-L1.
A diversity of IFN-regulated cytokines is elevated in the serum of patients with SLE versus HCs, in particular CXCL10 and CCL19, which have been shown to correlate with SLE disease severity and flares.6 In the analysis presented here, both CXCL10 and CCL19 were significantly downregulated by baricitinib 4 mg and were positively correlated with the IFN signature, anti-dsDNA titre and SLEDAI-2K overall disease activity. These findings, in addition to the effect on TNF superfamily members such as TNFRSF9, imply an indirect inhibition by baricitinib, and a potential role in lymphocyte migration, rather than only cellular activation and differentiation. Overall, these results suggest that lymphocyte recruitment, and migration into lymphoid organs and peripheral tissues, might be a unique potential mechanism of the action of baricitinib.
The patient population used in this analysis was primarily moderately active SLE patients, with a large representation of musculoskeletal and mucocutaneous manifestations and was comparable to other recent phase II–III clinical trials.
While the analysis presented here supports previous findings that baricitinib’s mechanism of action is partly mediated by its effects on the IFN signature, the other novel analytes identified are relevant to SLE immunopathogenic pathways such as B–T lymphocyte interactions, macrophage trafficking and signalling pathways linking the innate and adaptive arms of the immune system. The correlation between the expression of some of these molecules, like IL-6 and TNFRSF9, with clinical outcomes, in particular, joint manifestations and pain measures, expands not only our understanding of pathogenic mechanisms in SLE but also of potential response biomarkers.
Arthralgia in patients with SLE is mainly related to tenosynovitis as opposed to the erosion and joint destruction more common in rheumatoid and psoriatic arthritis.21 The correlation analysis presented here found no apparent relationship with IL-17 (the IL-12/–23 axis) but a potential link to the IL-6 pathway. Prior studies in rheumatoid arthritis analysing changes in cytokine production by B cells under IL-6R blockade22 found that B cell cytokine production did not simply suppress the IL-6 inflammatory axis but restored certain cytokines and chemokines under treatment, confirming a significant but weak correlation between IL-6 blockade and the production of macrophage inflammatory proteins and β-nerve growth factor at baseline, both relevant for persistent synovitis and pain sensation in rheumatoid arthritis. There are clear molecular (cytokine) differences related to joint manifestations in rheumatoid arthritis versus SLE, and the pathogenic mechanisms that lead to joint manifestations likely differ between the two diseases and remain to be fully delineated. However, lack of erosive changes and preferential tendon involvement in SLE versus rheumatoid arthritis suggest subtle pathogenic differences. The role of IL-6 in both diseases may also be different in terms of IL-17 induction.
IL-6 was also positively correlated with worst joint pain. It has been shown that IL-6 is an important modulator of pain through mechanisms that influence pain signalling at the central nervous system level. Specifically, for baricitinib, such an impact on pain has been demonstrated to happen beyond its impact on inflammation23 implying that, at least in part, baricitinib’s effect on pain is uncoupled from its anti-inflammatory mechanics.
Here, treatment with baricitinib 4 mg downregulated a potential network of cytokines involved in lymphocyte and monocyte/macrophage recruitment with CCL19, IFN-γ-induced proteins such as CXCL10 as well as TNFRSF9, IL-6 and others (TNF-α, IL-12β, PD-L1). However, mechanistically their impact on disease may differ since only CCL19, CXCL10, TNF-α and PD-L1 correlated with the IFN signature. Although CCL19 was positively correlated with SLEDAI-2K score and CXCL10 with anti-dsDNA titres, IL-6 and TNFRSF9 correlated with joint pain as well as swollen and tender joints. These results confirm previously reported data,11 and also broaden the evidence using an assay with sensitivity, extending the analysis at the protein level to additional IFN and non-IFN-related cytokines. The significant associations with clinical outcomes (although not at the highest level of correlation probably due to small sample size), in particular, for joint manifestations, pain and B/T cell activity, further expand our understanding of pathogenic mechanisms in SLE and identify potential response biomarkers for both systemic and organ-specific disease activity in patients with SLE.
However, recent inconclusive findings from two phase III clinical trials investigating the efficacy of baricitinib in patients with SLE (NCT03616912 and NCT03616964) pose a challenge to further interpret these biomarkers. A priori, this post hoc phase II analysis was powered for skin and joint manifestations but no other SLE domains (renal, haematology, central nervous system). Independent studies of the biomarkers identified by the current analysis will provide a rich resource to validate their impact comparing phase III responders and non-responders.
The downregulation of key cytokines and the observed correlations with clinical variables presented here indicate that treatment with baricitinib might be particularly effective in the subgroup of patients with high serological and disease activity (especially musculoskeletal manifestations). However, the recent inconclusive data from phase III trials warrant further analyses and consideration.
Of note, our study is limited to a preselected set of analytes (Olink INF I panel) and may have missed important analytes that are not part of the predefined inflammation assay panel. As well, while correlation analysis found the relationship between several analytes and clinical measures to be significant, the small number of patients from the phase II trial and the r values between 0.15 and 0.4 might further limit clinical relevance.
Despite the inconclusive results on the efficacy of baricitinib from the phase III trials, and the broad body of difficult to interpret literature already published on the topic of SLE pathogenesis and cytokine dysregulation, the analysis presented here remains relevant as it contributes to our understanding of the molecular pathways involved in SLE and the impact of baricitinib on immunological/cytokine signatures.
Conclusion
The analysis presented here advances the understanding of how baricitinib might act in patients with SLE by modulating multiple disease relevant proteins. However, based on inconclusive findings from the phase III trials, benefit of treatment might be limited. These results also serve to elucidate potential new biological targets that may impact SLE disease activity.
Data availability statement
Lilly provides access to all individual participant data collected during the trial, after anonymisation, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the US and EU and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.
Ethics statements
Patient consent for publication
Ethics approval
A full list of ERBs and approvals is attached as supplemental material. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We would like to thank the patients and investigators who participated in the study. Eli Lilly and Company or its representatives provided data, laboratory, and site monitoring services. Sample management assistance was provided by Mary Zuniga, BSc. Writing assistance was provided by Conor McVeigh, PhD, of Eli Lilly and Company.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
Contributors All authors contributed to the data analysis and interpretation, writing and critical revision of the publication, and final approval to submit, and were accountable for the accuracy and integrity of the publication. TD accepts full responsibility for the finished work and/or the conduct of the study, had access to the data and controlled the decision to publish.
Funding This study was sponsored by Eli Lilly and Company, under license from Incyte Corporation.
Competing interests TD has received grant support from Chugai, Janssen, Novartis and Sanofi, received consultancy support from AbbVie, Celgene, Eli Lilly and Company, Janssen, Novartis, Roche, Samsung and UCB, and speaker bureau fees from Eli Lilly and Company and Roche. YT has received speaking fees and/or honoraria from Gilead, AbbVie, Behringer-Ingelheim, Eli Lilly and Company, Mitsubishi-Tanabe, Chugai, Amgen, YL Biologics, Eisai, Astellas, Bristol-Myers, Astra-Zeneca, received research grants from Asahi-Kasei, AbbVie, Chugai, Mitsubishi-Tanabe, Eisai, Takeda, Corrona, Daiichi-Sankyo, Kowa, Behringer-Ingelheim, and consultant fees from Eli Lilly and Company, Daiichi-Sankyo, Taisho, Ayumi, Sanofi, GSK, AbbVie. MP has received grants and consulting fees from Eli Lilly and Company. ERD, AEK, MES, JART, JTS, ZS, and IDLT are employees and shareholders of Eli Lilly and Company.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.