Article Text

Abstract
Objective This phase 2a randomised, double blind, placebo controlled, parallel group study evaluated the safety and efficacy of a first-in-class drug candidate ABX464 (obefazimod, 50 mg and 100 mg per day), which upregulates the biogenesis of the mRNA inhibitor micro-RNA (miR)-124, in combination with methotrexate (MTX) in 60 patients (1:1:1 ratio) with moderate-to-severe active rheumatoid arthritis (RA) who have inadequate response to MTX or/and to an anti-tumour necrosis factor alpha (TNFα) therapy.
Methods The primary end point was the safety of ABX464; efficacy endpoints included the proportion of patients achieving American College of Rheumatology (ACR)20/50/70 responses, disease activity scores (DAS) 28, simplified disease activity score, clinical disease activity score), European League Against Rheumatism response, DAS28 low disease activity or remission.
Results ABX464 50 mg was safe and well tolerated. Two serious adverse events were reported (one on placebo group and one on ABX464 100 mg). Eleven patients were withdrawn for AEs (9 patients on 100 mg, 1 on 50 mg and 1 on placebo). Drug discontinuation was mainly due to gastrointestinal disorders. No cases of opportunistic infection, no malignancies and no death were reported. Compared with placebo, ABX464 50 mg showed significantly higher proportions of patients achieving ACR20 and ACR50 responses at week 12. DAS28-C reactive protein (CRP) and DAS28-erythrocyte sedimentation rate decreased significantly and rates of categorical DAS28-CRP response or CDAI remission increased significantly on ABX464 at week 12. A significant upregulation of miR-124 was observed in blood for every patient dosed with ABX464.
Conclusion ABX464 50 mg was safe, well tolerated and showed a promising efficacy. Mild-to-moderate gastrointestinal AEs led to a high drop-out rate of patients on ABX464 100 mg, which may not be a relevant dose to use. These findings warrant exploration of ABX464 at 50 mg per day or less for treating patients with RA.
Trial registration name Phase IIa randomised, double blind, placebo controlled, parallel group, multiple dose study on ABX464 in combination with MTX, in patients with moderate to severe active RA who have inadequate response to MTX or/and to an anti- TNFα therapy or intolerance to anti-TNFα therapy.
EUDRACT number: 2018-004677-27
Trial registration number NCT03813199.
- arthritis, rheumatoid
- methotrexate
- antirheumatic agents
Data availability statement
Data may be obtained from a third party and are not publicly available.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
ABX464 (obefazimod) is a first-in-class drug candidate for treating patients with active rheumatoid arthritis. ABX464 upregulates the biogenesis of the mRNA inhibitor micro-RNA-124 and can act as a natural brake on the production of various inflammatory mediators involved in inflammatory diseases.
WHAT THIS STUDY ADDS
This randomised, double-blind, placebo-controlled proof-of-concept study showed that at an oral daily dose of ABX464 50 mg was safe and well tolerated by patients.
An increased incidence of largely mild-to-moderate gastrointestinal adverse events in the ABX464 100 mg group led to a high drop-out rate of patients.
Several early efficacy endpoints showed promising results with ABX464 50 mg.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE AND/OR POLICY
Findings warrant further exploration of ABX464 as a rheumatoid arthritis treatment, using 50 mg per day or lower doses.
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease characterised by persistent synovitis and systemic inflammation. In the absence of disease control, RA ultimately results in severe progressive joint damage, disability, decreased quality of life, comorbidities and higher risk of mortality.1 2 Conventional therapy with synthetic disease-modifying antirheumatic drugs (DMARDs) should be started as soon as the diagnosis of RA is made and methotrexate (MTX) should be part of the first treatment strategy.2 If treatment target is not achieved and if poor prognosis factors are present, a targeted therapy (biological (b) or targeted synthetic (ts) DMARD) is added. Although ts- and bDMARD drugs have revolutionised RA prognosis, safety concerns and difficult-to-treat RA3 4 leave a room for novel, safe and effective oral targeted treatments, which may act with a different mechanism of action and better control the course of the disease.
ABX464 (obefazimod) upregulates the biogenesis of the mRNA inhibitor micro-RNA (miR)-124, which in turn modulates monocyte and macrophage activations5–8 and can act as a natural brake on the production of various inflammatory mediators involved in inflammatory diseases such as ulcerative colitis (UC) and RA.5–10 Among targets that miR-124 represses, CCL2 (monocyte chemoattractant protein-1, MCP-1), Signal transducer and activator of transcription 3 (STAT3), interleukine 6 receptor and tumour necrosis factor alpha (TNFα) converting enzyme (TACE) are important actors in RA.11–13 ABX464 has demonstrated durable efficacy in treating patients suffering from UC, a chronic inflammatory disorder of the colonic mucosa.14 The efficacy and safety profiles of this compound are likely related to its unique mechanism of action.15 Recently, non-clinical in vivo data provided evidence that ABX464 (40 mg/Kg, per os) strongly reduced the incidence of collagen-induced arthritis in DBA-1 male mice.10 These encouraging data have led to design the present 12-week randomised double-blind, placebo-controlled proof of concept study aimed to investigate the safety of ABX464 administered daily (50 mg or 100 mg) in patients with RA with active disease not responding to MTX.
Method
The study was conducted in 21 centres (eight in France, three in Belgium, seven in Poland and three in Hungary) in accordance with the standard operating procedures of the sponsor, which were designed to ensure adherence with the Declaration of Helsinki and Good Clinical Practice.
Patients
Eligible patients (ages 18–75 years) had a confirmed and documented diagnosis of RA, for at least 12 weeks, according to the revised 2010 American College of Rheumatology (ACR)-European League Against Rheumatism (EULAR) classification criteria, in addition to at least one positive criteria among rheumatoid factor, anticitrullinated peptide antibody or bone erosion. Inclusion criteria comprised swollen joint count (SJC) of ≥4 (28-joint count) and tender joint count of ≥4 (28-joint count), disease activity score (DAS)28 C reactive protein (CRP) of ≥3.2 and CRP ≥5 mg/L. Patients either had an inadequate response or failed either MTX (≥10 mg/week) or/and anti-TNFα therapy or were intolerant to anti-TNFα therapy for ≥12 weeks before trial entry. Exclusion criteria comprised a confirmed diagnosis of systemic lupus erythematosus or active or history of serious or opportunistic infections (OIs). Patients with a history of immunodeficiency or with history of malignancy were also excluded. In addition, patients were excluded if previously treated with non-anti-TNF bDMARDs, tsDMARDs, systemic corticosteroids at a dose >10 mg/day within 2–4 weeks prior to the study; or immunosuppressive drugs.
All participants provided written informed consent before participation. First patient was randomised on 1 August 2019 and the last patient was randomised on 2 February 2021; the last patient completed on 27 April 2021.
Randomisation and procedures
Randomisation was performed via a centralised treatment allocation system. Eligible patients were randomised according to a 1:1:1 ratio and were treated for 12 weeks followed by a 21-day follow-up period. During the treatment phase, patients were receiving either capsules containing ABX464 50 mg (n=21), ABX464 100 mg (n=19) or placebo (n=20) given orally once per day in a fed condition. Patients who completed the induction study at week 12 could roll over into a 52-week open-label maintenance study to evaluate the long-term safety and efficacy of 50 mg per day oral ABX464 in RA (trial ABX464-302; NCT04049448). Here, we present the results of the 12-week treatment induction phase of this clinical trial.
Assessments
Safety
The primary end point was the safety and tolerability of two doses of ABX464 versus placebo. The primary safety endpoint was the rate of all treatment-emergent adverse event (TEAEs). Safety was evaluated based on adverse events (AEs), clinical laboratory parameters, vital signs, physical examination, standard 12-lead ECG results. AEs were coded using the Medical Dictionary for Regulatory Activities (V.23.0). Severity of AEs was graded on a 5-point scale according to the Common Terminology Criteria for Adverse Events (V.5.0). The causal relationship between the study treatment and the occurrence of each AE was assessed by each investigator using clinical judgement.
Efficacy
Efficacy endpoints included an evaluation of the effects of ABX464 versus placebo on (1) the ACR 20/50/70 responses (≥20/50/70% improvement in tender/painful joint count (TJC) (28-joint count) and SJC (28-joint count) plus ≥20/50/70% improvement in three of the five remaining ACR core set measures) and all, components of the ACR response (CRP, TJC, SJC, pain-patient assessment of joint pain, patient global assessment of disease (PtGA), physician’s global assessment of disease, disability index of the healthy assessment questionnaire (HAQ-DI), (2) DAS28 scores, simplified disease activity score (SDAI) and clinical disease activity score (CDAI), (3) clinical response (DAS28 EULAR good and moderate responses), (4) low disease activity (LDA; DAS28-erythrocyte sedimentation rate (ESR) ≤3.2) or remission (DAS28-ESR remission (DAS28 <2.6), ACR/EULAR Boolean remission (TJC(28), SJC(28), CRP (mg/dL) and PtGA, all ≤1), SDAI remission (SDAI ≤3.3) and CDAI remission (CDAI ≤2.8), (5) Functional Assessment of Chronic Illness Therapy (FACIT)-Fatigue score.
Laboratory tests
Blood samples from patients in each group were used to measure the expression of miR−124 at baseline and week 8. For each sample, RNA and DNA were extracted (AllPrep DNA/RNA/miRNA Universal kit, Qiagen) and their concentrations were determined (Nanodrop 2000 and Qubit V.2.0 fluorometer, ThermoFisher). The miRNAs were retro transcribed from 70 ng of the extracted RNA (TaqMan Advanced miRNA cDNA Synthesis Kit, ThermoFisher), then, after preamplification, cDNA was subjected to duplicate droplet digital PCR measurements using two singleplex assays targeting miR-16 and miR-124 at specified dilutions (1:10 for miR-124; 112 000 for miR-16).
Statistical methods
The rate of all TEAEs, categorical DAS28-CRP response, DAS28-ESR remission, SDAI remission, CDAI remission, ACR/EULAR Boolean remission, LDA and ACR 20/50/70 response rates were compared in patients who received a dose of ABX464 or placebo by likelihood ratio χ2 test on a 10% two-sided significance level. A mixed model analysis of covariance was conducted for change from baseline in SDAI and CDAI scores, DAS28-CRP, DAS28-ESR, ESR, CRP and all ACR components.
The Full Analysis dataset (FAS population) was defined as those patients included in the study, who had received at least one dose of the study treatment, and who had at least one baseline data. Post hoc efficacy analyses were performed for the intent-to-treat (ITT, ie, patients randomised, regardless of whether the patient received a dose of study treatment or completed the study) and per-protocol (PP) (patients of the FAS population without any major protocol deviation) populations. Additional post hoc efficacy and safety analyses were also performed on subgroups of patients with or without previous exposure to anti TNFα therapy.
Statistical analyses were carried out using SAS, V.9.4 or later (SAS Institute, Cary, North Carolina). For miR-124 analyses, non-parametric Kruskal-Wallis test followed by a Dunn’s multiple comparisons test was performed using GraphPad Prism software (V.9.3.0).
Results
Demographics and baseline characteristics
Patient disposition is summarised in figure 1. For FAS, ITT and Safety sets, there were 19, 21 and 20 patients in ABX464 100 mg, 50 mg and placebo groups, respectively. For PP set, there were 18, 20 and 20 patients in ABX464 100 mg, 50 mg and placebo groups, respectively. Baseline demographic characteristics were similar among the three groups (table 1). Most patients (61.7%) were women, with a mean age of 57.0 years and a mean RA duration of 6.4 years. Thirty per cent of patients were previously exposed to anti-TNF therapies and 53.3% received corticosteroids at baseline. At baseline, mean DAS28-CRP and DAS28-ESR were, respectively, 5.4 and 5.8. History of MXT dose was comparable across treatment groups.
Baseline patient demographic and clinical characteristics (Full Analysis set*)
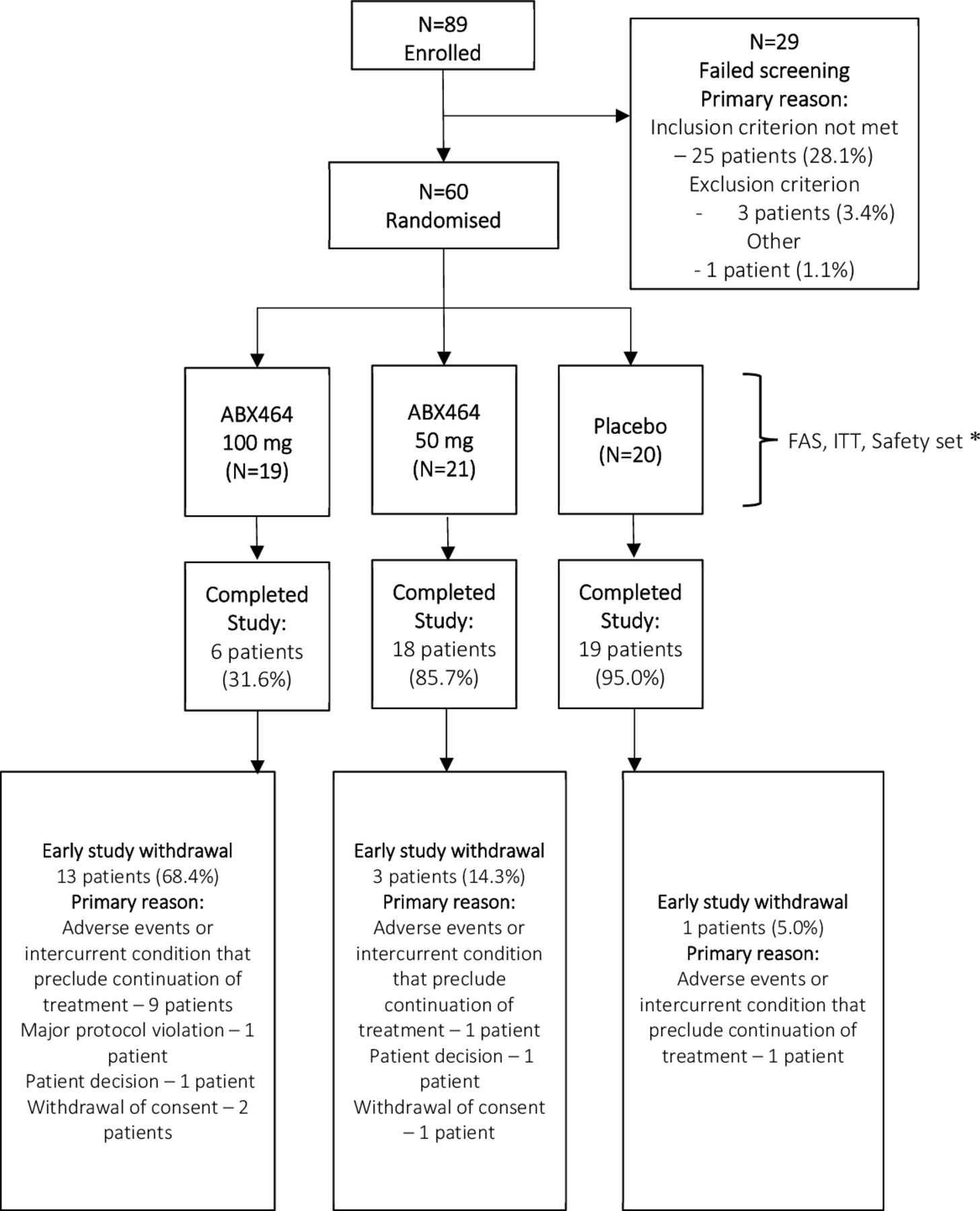
Patient disposition. *: For PP set, there were 18, 20 and 20 patients in ABX464 100 mg, 50 mg and placebo groups, respectively. To derive the PP set, one patient was excluded in the ABX464 100 mg group, one patient was excluded in the ABX464 50 mg and no patient was excluded in the placebo group. FAS, Full Analysis dataset; ITT, intent-to-treat.
Safety
A total of 220 AEs were reported by 50 subjects (83.3%) during the study, and among them, 211 AEs were treatment emergent. Treatment with ABX464 was associated with the occurrence of mainly mild to moderate AEs. AEs occurred more frequently in the ABX464 treatment groups than in the placebo group (AEs, 94.7%, 85.7% and 70.0%, in ABX464 100 mg, 50 mg and placebo groups, respectively). When compared with placebo, a higher incidence of TEAEs was reported in the ABX464 groups, with a statistically significant difference for the 100 mg dose group (p=0.035) (online supplemental table 1). The onsets of headaches, nausea and vomiting were more rapid (mostly in the first 2 weeks) than the onset of diarrhoea (mostly in the two first months). Most dropouts in the 100 mg group took place rapidly in the first 2 weeks of treatment (online supplemental figure 1). One serious AE (SAE) was reported in each treatment group, including one atrial fibrillation in the 100 mg ABX464 group and a severe COVID-19 disease in the placebo group, of which both were treatment emergent. The patient hospitalised for a severe atrial fibrillation had experienced severe diarrhoea and the conclusion of hospitalisation was an atrial fibrillation reactive to hypokalemia secondary to diarrhoea in the context of study drug treatment. Another SAE was a severe RA worsening, as it required patient hospitalisation; it occurred before the first ABX464 50 mg dose. The incidence of severe AEs (grade 3 or above) was numerically higher in the ABX464 50 mg and 100 mg groups (14.3% and 15.8%, respectively) than in placebo group (5%).
Supplemental material
Supplemental material
TEAEs that occurred in >5% of patients in any ABX464 treatment group included headache (22 patients, 36.7%), followed by diarrhoea (12 patients, 20.0%), nausea (12 patients, 20.0%), abdominal pain upper (10 patients, 16.7%), vomiting (5 patients, 8.3%), dyspepsia (4 patients, 6.7%) and RA exacerbation (4 patients, 6.7%). Except for abdominal pain and RA exacerbation, the highest incidence of TEAEs occurred in the 100 mg ABX464 group (table 2). In the 100 mg ABX464 group, there were numerically higher incidences for neurological (dizziness, headache, taste disorder, tremor) and infectious AEs than for ABX464 50 mg and placebo groups. Drug discontinuation was mainly due to gastrointestinal disorders (online supplemental table 2) and was the main cause of patient withdrawals. Eleven patients were withdrawn for AEs (nine patients in the 100 mg ABX464 group and one in each of the other groups). Nine patients had temporary discontinuation (four patients in each of the ABX464 groups and one patient in the placebo group) with a mean duration of 4 days (ranging from 1 to 9 days). Vital signs and ECG parameters were similar across the ABX464 50 mg and 100 mg groups and the placebo group. No clinically meaningful changes were observed in laboratory data or other safety assessments (online supplemental table 3). No cases of OI were recorded, and the infestation rate was similar between placebo and ABX464 groups. No deaths or malignancies were reported. No AE persisted or had sequelae at the end of trial.
Most commonly occurring TEAEs by system organ class (>5% patients) and preferred term (Safety Set*)
Efficacy
In the ITT population, the DAS28-CRP and DAS28-ESR decreased sequentially from baseline to week 12 after both ABX464 doses. Decreases in the DAS28-CRP and DAS28-ESR were statistically significant between the placebo group and the 50 mg ABX464 dose at week 12 (p=0.043 and p=0.035, respectively) (figure 2, table 3). In the PP population, significant decreases in the DAS28-CRP and DAS28-ESR were seen between placebo and the 50 mg or 100 mg ABX464 groups as early as week 8 (figure 2, online supplemental table 4).

{kind=link}
{kind=link}
DAS28-CRP (A) and DAS28-ESR (B) mean (±SEM) changes from baseline at weeks 4, 8 and 12 in RA patients who received placebo or ABX464 (50 or 100 mg) once daily (ITT set). ITT, intent-to-treat; RA, rheumatoid arthritis.
Changes from baseline at week 12 in ESR, DAS28-CRP, DAS28-ESR, SDAI, CDAI, SJC, TJC, CRP, Pain-VAS, HAQ-DI and FACIT-Fatigue (ITT set)
Compared with placebo, mean changes in CDAI scores from baseline to week 12 were significantly different for the 50 mg ABX464 dose in the ITT population (p=0.020) (table 3) and for both ABX464 doses in the PP population (online supplemental table 4). No significant differences between groups were observed for SDAI scores change in ITT and PP populations.
Rates of categorical DAS28-CRP response at week 12 were significantly higher versus placebo at 50 mg and 100 mg ABX464 doses in the PP population (p=0.004 and p=0.038, respectively) (online supplemental table 5). These differences did not reach statistical significance in the ITT population (table 4).
Patients’ responses and remissions at week 12 (ITT set)
In the ITT and PP populations, difference in LDA responses across groups did not reach statistical significance at week 12 (table 4, online supplemental table 5). There was a significant difference in CDAI remission rates at week 12 between the ABX464 50 mg group and the placebo group (ITT set: p=0.039; PP set: p=0.024). DAS28-ESR remission, SDAI remission and ACR/EULAR remission rates were not significantly different across groups (table 4, (online supplemental table 5).
In the ITT population, differences between treatment groups for the proportions of patients achieving ACR20, ACR50 and ACR50 did not reach statistical significance (table 4). When considering the very small-sized subsets of patients exposed or not to anti-TNFα, ACR20, ACR50 and ACR70 response rates at week 12 were not significantly different across groups (online supplemental table 6). In the PP population, the proportions of patients achieving ACR20 and ACR50 responses at week 12 were significantly higher (p=0.030 and p=0.037, respectively) in the ABX464 50 mg group than in placebo (online supplemental table 5). The ACR20 and ACR50 response rates were numerically higher in patients receiving ABX464 100 mg versus placebo, but statistical significance was not reached. ACR70 response rates and mean changes in HAQ-DI were not significantly different across groups at week 12. There was a significant difference in change in FACIT-Fatigue at week 12 between the ABX464 50 mg group and the placebo group in PP analysis (p=0.045, online supplemental table 4).
Two efficacy sets were initially considered: the ‘raw’ efficacy set 1 took into consideration only patients at time point (N=number of patients from whom efficacy variables were available at week 12). The efficacy set 2 used data imputation (last observation carried forward) from week 4 onwards. If there were no efficacy data available, then the dropout was considered as treatment failure. Main data obtained in the efficacy set 2 are provided in online supplemental tables 7 and 8.
The pharmacokinetics (PK) analysis was performed on the PK Analysis Set, which included all the patients who received at least one dose of ABX464 and for whom postdose concentrations were available without major protocol deviations or events implying bias for the PK evaluation. Summary of PK parameters following administration of ABX464 50 mg and 100 mg is provided in online supplemental table 9.
miR-124
The expression of miR-124 was measured in 27 blood samples from patients with RA and a miR-124 induction was observed at week 8 compared with baseline in patients receiving ABX464. The fold changes of miR-124 were statistically significant between placebo and ABX464 50 and 100 mg doses (p<0.001 and p<0.01, respectively) with medians equal to 0.2 for placebo, 174.5 and 119.2 for ABX464 50 and 100 mg, respectively (online supplemental figure 2).
Supplemental material
Discussion
This is the first multicentre, randomised, double-blind, placebo-controlled study to evaluate the safety and efficacy of two daily doses (50 mg and 100 mg) of ABX464 (obefazimod) for 12 weeks in patients with moderate to severe active RA who had an inadequate response to MTX and/or to an anti-TNFα therapy.
A daily dose of 50 mg of ABX464 appeared safe in those patients and no new safety signals were identified. ABX464 AEs were mild, dose-dependent, and the nature of these AEs was consistent with what has been observed in more than 850 subjects who have so far been treated in other clinical trials with ABX464 across different indications (HIV/AIDS, COVID-19 and UC). Compared with placebo group, a larger proportion of patients in ABX464 groups experienced TEAEs. An increased incidence of largely mild-to-moderate gastrointestinal AEs in the 100 mg treatment group led early to a high drop-out rate of patients, and, therefore, may not be a relevant dose to use. Rates of AEs—in particular, abdominal pain upper, diarrhoea, vomiting and headache—were more elevated than rates of AEs observed throughout the development programme of ABX464 with the dose 50 mg and 100 mg16; these higher rates were likely driven by an additive effect due the concomitant administration of MTX since gastrointestinal AEs are the most common side effect with this treatment.17 The ABX464 100 mg group seems to have higher incidences not only for gastrointestinal but also for neurological (dizziness, headache, taste disorder, tremor) and infectious AEs; the ABX464 100 mg/day dose is probably too high and the future development programme will explore doses lower than 50 mg. In the present study, no malignancies were reported, and no OI was observed during the study, with an infestation rate similar between placebo and ABX464 all doses.
Although the sample size of this proof-of-concept study was limited, multiple early efficacy endpoints showed signs of promise with the ABX464 50 mg daily dose, whereas no clear efficacy was demonstrated with the highest dose, likely due to the high drop-out rate of patients. In the ITT population, DAS28-CRP and DAS28-ESR of ABX464 50 mg-treated patients decreased sequentially to reach a statistically significant difference versus placebo after 12 weeks. In addition, compared with placebo, changes in CDAI scores from baseline to week 12 in the ITT population were significantly in favour of ABX464 50 mg, as were differences in CDAI remission rates. Decrease in CRP levels did not reach significance, presumably due to a very large SD. In general, production of CRP correlates with IL-6 levels, which were significantly decreased in the serum of patients with RA (data not shown). Significant changes were likewise observed in the PP population, with additional endpoints in favour of ABX464, such as categorical DAS28-CRP response rates and ACR20 and ACR50. In the PP set, more than half of ABX464 patients dosed with 50 mg reached the ACR20 endpoint at week 12.
From a mechanism of action standpoint, a striking increase in miR-124 expression was seen at week 8 in blood samples of patients with RA receiving ABX464, which indicates that the treatment promotes the production of a key agent that has the potential to reduce several inflammatory-activated pathways. Indeed, miR-124 target genes that control the production of a number of inflammatory mediators (eg, TNF-α, IL-6, IL-17) implicated in the intense inflammatory reaction that drives RA.5–9 An upregulated biogenesis of miR-124 has been consistently reported with ABX464, across non-clinical and clinical studies in different indications,15 still a definitive correlation with clinical response remains to be established. Interestingly though, no significant difference in miR-124 upregulation was seen between the two ABX464 doses, indicating the absence of a dose-dependent effect, which reminds the absence of dose-related efficacy of the drug reported in clinics in patients with UC.14
In conclusion, this proof-of-concept study provides the first clinical evidence that ABX464 50 mg/day for 12 weeks appeared to be safe and well tolerated by patients with active RA, whereas 100 mg may not be a relevant dose to use in patients with RA. Despite a limited number of patients per group, multiple efficacy endpoints showed promising results with the ABX464 50 mg/day. A total of 40 patients from the present cohort have been enrolled in an ongoing open-label 52-week maintenance study with ABX464 50 mg per day to collect further safety and efficacy data. Though preliminary, these encouraging findings warrant further exploration of the efficacy and safety of ABX464 at 50 mg per day or less as an RA treatment.
Data availability statement
Data may be obtained from a third party and are not publicly available.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Federal Agency for Medicines and Health ProductsEurostation II—Place Victor Horta 40/401060 BrusselsState Institute For Drug Control (SUKL) Srobarova 48 10041 Praha 10, CZANSM—143/147, bd Anatole France—93285 Saint-Denis Cedex, FR OGYÉI/33889-6/2019 1051 Budapest, Zrínyi utca 3.Levélcím: 1372 Postafiók 450, HNClinmark ul. Wiktorska 63 02-587 Warszawa, PL. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors would like to thank all other investigators participating to the study: D. Chudzik, G. Cormier, D. Cornec, S. Daniluk, K. Fazekas, P. Goupille, M. Korkosz, B. Kwiatkowska, P. Leszczyński, B. Rojkovich, C. Roux, C. Salliot, F. Van den Bosch, T. Varga, P. Verschueren. The authors would like to thank P.A. Boyer on behalf of Abivax, for his assistance in writing and preparing the manuscript.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
Contributors All authors contributed equally to the conception and development of the manuscript, including literature review. The final version was approved by all authors. The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). HE is the guarantor.
Funding This study was supported by Abivax. The authors received no direct compensation related to the development of this manuscript.
Competing interests CD received consulting fees from ABIVAX as PI of the study, and declares consulting fees, punctual links or research grant from Abbvie, Amgen, BMS, Fresenius-Kabi, MSD, Novartis, Pfizer, Sandoz, Sanofi, Roche-Chugai, UCB. PD and MK were investigators contracted by the Sponsor for this study and have nothing else to disclose. LDdR, PG, HE, SB, DS and JS are employees at Abivax. JMS was a former employee at Abivax.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.