Article Text

Abstract
Objectives In the anifrolumab systemic lupus erythematosus (SLE) trial programme, there was one trial (TULIP-1) in which BILAG-based Composite Lupus Assessment (BICLA) responses favoured anifrolumab over placebo, but the SLE Responder Index (SRI(4)) treatment difference was not significant. We investigated the degree of concordance between BICLA and SRI(4) across anifrolumab trials in order to better understand drivers of discrepant SLE trial results.
Methods TULIP-1, TULIP-2 (both phase 3) and MUSE (phase 2b) were randomised, 52-week trials of intravenous anifrolumab (300 mg every 4 weeks, 48 weeks; TULIP-1/TULIP-2: n=180; MUSE: n=99) or placebo (TULIP-1: n=184, TULIP-2: n=182; MUSE: n=102). Week 52 BICLA and SRI(4) outcomes were assessed for each patient.
Results Most patients (78%–85%) had concordant BICLA and SRI(4) outcomes (Cohen’s Kappa 0.6–0.7, nominal p<0.001). Dual BICLA/SRI(4) response rates favoured anifrolumab over placebo in TULIP-1, TULIP-2 and MUSE (all nominal p≤0.004). A discordant TULIP-1 BICLA non-responder/SRI(4) responder subgroup was identified (40/364, 11% of TULIP-1 population), comprising more patients receiving placebo (n=28) than anifrolumab (n=12). In this subgroup, placebo-treated patients had lower baseline disease activity, joint counts and glucocorticoid tapering rates, and more placebo-treated patients had arthritis response than anifrolumab-treated patients.
Conclusions Across trials, most patients had concordant BICLA/SRI(4) outcomes and dual BICLA/SRI(4) responses favoured anifrolumab. A BICLA non-responder/SRI(4) responder subgroup was identified where imbalances of key factors driving the BICLA/SRI(4) discordance (disease activity, glucocorticoid taper) disproportionately favoured the TULIP-1 placebo group. Careful attention to baseline disease activity and monitoring glucocorticoid taper variation will be essential in future SLE trials.
Trial registration numbers NCT02446912 and NCT02446899.
- Autoimmune Diseases
- Lupus Erythematosus, Systemic
- Biological Therapy
Data availability statement
Data are available on reasonable request. Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Disclaimer: this video summarises a scientific article published by BMJ Publishing Group Limited (BMJ). The content of this video has not been peer-reviewed and does not constitute medical advice. Any opinions expressed are solely those of the contributors. Viewers should be aware that professionals in the field may have different opinions. BMJ does not endorse any opinions expressed or recommendations discussed. Viewers should not use the content of the video as the basis for any medical treatment. BMJ disclaims all liability and responsibility arising from any reliance placed on the content.
WHAT IS ALREADY KNOWN ON THIS TOPIC
In the anifrolumab clinical development programme for patients with systemic lupus erythematosus (SLE), clinical efficacy outcomes in favour of anifrolumab were consistently observed across the composite endpoints BILAG-based Composite Lupus Assessment (BICLA) and SLE Responder Index (SRI(4)) in the phase 3 TULIP-2 trial and the phase 2b MUSE trial, but not in the phase 3 TULIP-1 trial, for which SRI(4) treatment differences did not reach statistical significance.
WHAT THIS STUDY ADDS
Assessment of BICLA and SRI(4) outcomes at an individual patient level across TULIP-1, TULIP-2 and MUSE identified a high level of concordance between both composite endpoints, and higher proportions of patients met both BICLA and SRI(4) response definitions (‘dual responders’) with anifrolumab than placebo.
A discordant BICLA non-responder/SRI(4) responder subgroup was identified in all three trials, but this subgroup was over-represented in the placebo group of TULIP-1, which resulted in a reduction in the magnitude of the overall TULIP-1 SRI(4) treatment effect.
In this discordant TULIP-1 subgroup, placebo-treated patients had lower baseline disease activity, joint counts and glucocorticoid tapering rates than anifrolumab-treated patients, which may have contributed to more placebo-treated patients with SLE Disease Activity Index 2000 (SLEDAI-2K) arthritis responses.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE AND/OR POLICY
Given the array of challenges posed by SLE clinical trials, investigators and regulators need to understand why endpoints are not attained or are discordant; our analysis has lessons for all investigators involved in SLE clinical trials, and we recommend careful attention to baseline disease activity and minimising glucocorticoid taper variation in future SLE trials.
Introduction
Systemic lupus erythematosus (SLE) is a heterogeneous autoimmune disease that can affect multiple organ systems and causes substantial disease burden.1–4 As standard therapies do not always adequately control disease activity, additional effective SLE-targeted therapies are needed, which has led to unprecedented SLE clinical trial activity over the last two decades. Efficacy assessments in these trials often use composite indices of global lupus disease activity, such as the BILAG-based Composite Lupus Assessment (BICLA) and the SLE Responder Index (SRI).5–7
Anifrolumab is a human monoclonal antibody to the type I interferon receptor that is approved in the United States, Japan and Canada for the treatment of adult patients with moderate to severe SLE receiving standard therapy.8–10 Anifrolumab was investigated in patients with SLE in the phase 2b MUSE trial and in the phase 3 TULIP-1 and TULIP-2 trials.11–13 Clinical responses were assessed using both the BICLA and SRI(4) composite indices.11–13 MUSE had an SRI(4)-based primary endpoint and, given the robust outcomes, SRI(4) was originally selected as the primary endpoint for TULIP-1 and TULIP-2. BICLA, a secondary endpoint that also yielded robust outcomes in MUSE and TULIP-1, was subsequently designated the primary endpoint in TULIP-2 prior to unblinding of the TULIP-2 dataset.14 In TULIP-2, anifrolumab demonstrated a statistically significant benefit compared with placebo measured by both BICLA and SRI(4) responses at week 52; similar results were also observed in MUSE.11–14 In TULIP-1, the effect of anifrolumab 300 mg on BICLA response was of similar magnitude to that seen in TULIP-2 and MUSE; however, the treatment difference between anifrolumab and placebo with SRI(4) did not achieve statistical significance.
While BICLA and SRI(4) both evaluate clinically meaningful elements of global SLE disease activity,15 differences in their metric properties may give rise to inconsistent classification of a patient’s response between these measures.16 The BILAG-2004 index, on which the BICLA is anchored, grades each manifestation according to severity and the physician’s intention to treat; it also captures incremental, clinically meaningful improvement or worsening.7 17 18 By contrast, the Systemic Lupus Erythematosus Disease Activity Index 2000 (SLEDAI-2K), on which the SRI(4) is anchored, consists of dichotomous scoring (present/absent) of each manifestation independent of severity and assigns differential weights to the SLEDAI-2K elements.5 19 20 To be a BICLA responder, a patient must have at least partial improvement in all severe (BILAG-2004 A) or moderate (BILAG-2004 B) clinical manifestations affected at baseline, whereas to be an SRI(4) responder, a patient needs to have complete resolution of enough manifestations affected at baseline to reduce the total SLEDAI-2K score by ≥4 points.17
In this analysis, we investigated the degree of concordance between BICLA and SRI(4) across anifrolumab trials to better understand drivers of discrepant SLE trial results. In particular, we aimed to determine whether a subgroup of patients with discordant BICLA and SRI(4) outcomes could be identified that may explain the lack of significant SRI(4) treatment difference in TULIP-1 and, more generally, whether we could draw lessons to inform future SLE trial design/execution.
Patients and methods
Patients and Study Design
Detailed methods for TULIP-1, TULIP-2 and MUSE have been reported.11–13 TULIP-1, TULIP-2 and MUSE were randomised, double-blind, 52-week trials of adult patients with autoantibody positive moderate to severe SLE receiving standard therapy. Here we analysed data from patients who received the target dose of anifrolumab 300 mg for 48 weeks) or placebo.
In TULIP-1 and TULIP-2, attempts to taper oral glucocorticoids to ≤7.5 mg/day (prednisone or equivalent) were mandatory between weeks 8 and 40 for patients receiving ≥10 mg/day at baseline, and taper was considered sustained if maintained from weeks 40 to 52. In MUSE, oral glucocorticoid tapering was encouraged for all patients but was at the discretion of investigators.
BICLA and SRI(4) Endpoints
The TULIP-1, TULIP-2 and MUSE trials included analyses of BICLA and SRI(4) responses at week 52. A BICLA response was defined as all of the following: reduction of all baseline BILAG-2004 A domain scores to B/C/D, and all B domain scores to C/D, and no worsening in other BILAG-2004 organ systems as defined by ≥1 new BILAG-2004 A or ≥2 new BILAG-2004 B domain scores; no increase in SLEDAI-2K score (from baseline); no increase in Physician’s Global Assessment (PGA) score (≥0.3 points from baseline); no use of restricted medications beyond protocol-allowed thresholds; and no discontinuation of investigational product. An SRI(4) response was defined as all of the following:≥4-point reduction in SLEDAI-2K; <1 new BILAG-2004 A or <2 new BILAG-2004 B organ domain scores; no increase in PGA (≥0.3 points from baseline); no use of restricted medications beyond protocol-allowed thresholds; and no discontinuation of investigational product.
Assessment of Concordance on BICLA and SRI(4) Outcomes
In TULIP-1, TULIP-2 and MUSE, assessments of BICLA and SRI(4) responses at week 52 were performed for all patients who received anifrolumab 300 mg or placebo. Patients were grouped by concordance on BICLA and SRI(4) outcomes. Concordant subgroups included patients who were both BICLA and SRI(4) responders (‘dual’ responders), or patients who were both BICLA and SRI(4) non-responders. Discordant subgroups included patients who were BICLA non-responders and SRI(4) responders, or BICLA responders and SRI(4) non-responders.
Concordant and discordant subgroups were evaluated for baseline demographics and clinical characteristics, glucocorticoid taper, responses from baseline to week 52 across the BILAG-2004 and SLEDAI-2K organ domains, and joint count changes from baseline to week 52.
Statistical Analyses
The proportion (and corresponding treatment differences, 95% CIs, and nominal p values) of patients achieving a dual BICLA and SRI(4) response was compared in the anifrolumab vs placebo groups using a Cochran-Mantel-Haenszel approach controlling for stratification factors (SLEDAI-2K score at screening (<10/≥10), glucocorticoid daily dose on day 1 (<10/≥10 mg/day) and type I interferon gene signature at screening (high/low)).21 Percentage agreement and Cohen’s kappa were used as measures of agreement between BICLA and SRI(4) responses in each study. The percentage agreement was calculated as the number of agreement scores divided by the total number of scores (percentage agreement in MUSE was based on all patients with ≥1 BILAG-2004 A or B score at baseline). The Cohen’s kappa coefficient (ĸ, defined as the amount by which the observed agreement exceeds that expected by chance alone, divided by the theoretical maximum22) was used to evaluate the degree of concordance/reliability between the two endpoints; ĸ<0 is ‘no agreement,’ ĸ=0–0.20 is ‘slight agreement,’ ĸ=0.21–0.40 is ‘fair agreement,’ ĸ=0.41–0.60 is ‘moderate agreement,’ ĸ=0.61–0.80 is ‘substantial agreement’ and ĸ=0.81–1.0 is ‘perfect agreement’.23
Patient and Public Involvement
Patients and/or the public were not involved in the design, conduct, reporting, or dissemination of this research.
Results
Patients
The anifrolumab 300 mg and placebo groups in TULIP-1 (anifrolumab, n=180; placebo, n=184), TULIP-2 (anifrolumab, n=180; placebo, n=182) and MUSE (anifrolumab, n=99; placebo, n=102) were assessed. Patient demographics and clinical characteristics were generally balanced across treatment groups, both within the individual trials, and across TULIP-1, TULIP-2 and MUSE (online supplemental table S1). Most patients (>91% in all groups) were female. At baseline, the mean SLEDAI-2K scores ranged from 10.7 to 11.5, and approximately half of all treatment groups had ≥1 BILAG-2004 A score (45.0%–52.5%). Across treatment groups, 78.3%–86.3% of patients were receiving oral glucocorticoids at any dose, and 45.6%–62.7% were receiving glucocorticoids ≥10 mg/day.
Supplemental material
BICLA and SRI(4) Concordance
The concordance between BICLA and SRI(4) responder status at week 52 for patients in TULIP-1, TULIP-2 and MUSE is summarised in figure 1. Across the three trials, 85.4% (TULIP-1), 83.7% (TULIP-2) and 78.0% (MUSE) of patients had concordant BICLA and SRI(4) outcomes. The Cohen’s kappa analysis showed substantial agreement between the outcomes (TULIP-1 and TULIP-2: ĸ=0.7, nominal p<0.001; MUSE: ĸ=0.6, nominal p<0.001).

Concordance between patient responder status for BICLA and SRI(4) outcomes at week 52 in TULIP-1, TULIP-2 and MUSE (%). BICLA, British Isles Lupus Assessment Group-based Composite Lupus Assessment; SRI(4), Systemic Lupus Erythematosus Responder Index of ≥4.
In TULIP-1, the proportions of patients who were both BICLA and SRI(4) responders (‘dual’ responders) were 42.2% for the anifrolumab group and 27.7% for the placebo group (figure 1). This treatment difference was statistically significant (14.3%; 95% CI 4.6% to 24.0%; nominal p=0.004), and was consistent with differences observed in TULIP-2 (16.9%; 95% CI 7.2% to 26.7%; nominal p<0.001) and in MUSE (27.7%: 95% CI 15.7% to 41.5%; nominal p<0.001).
BICLA and SRI(4) Discordance
Smaller proportions of patients in each study had discordant BICLA and SRI(4) outcomes (figure 1). In TULIP-2 and MUSE, the patterns of discordance were generally similar across the treatment groups. In TULIP-1, however, more patients in the placebo group (n=28, 15.2%) than the anifrolumab 300 mg group (n=12, 6.7%) were BICLA non-responders/SRI(4) responders; thus, the placebo group reduced the overall TULIP-1 SRI(4) treatment effect by −8.5 percentage points. This subgroup constituted 11.0% (n=40) of the TULIP-1 population.
Demographics, Clinical Characteristics and Glucocorticoid Use in the TULIP-1 BICLA Non-responder/SRI(4) Responder Subgroup
Patient demographics were similar across the BICLA non-responder/SRI(4) responder subgroup, concordant subgroups and the overall TULIP-1 population (online supplemental table S2). In TULIP-1, a greater proportion of patients receiving placebo compared with anifrolumab were from Eastern Europe (38.0% vs 28.9%), and this difference was most conspicuous in the BICLA non-responder/SRI(4) responder subgroup (15/28 (53.6%) vs 3/12 (25.0%)).
Among patients in the TULIP-1 BICLA non-responder/SRI(4) responder subgroup, those who received placebo had lower baseline SLEDAI-2K scores and joint counts than those who received anifrolumab (table 1). The placebo group also had a smaller proportion of patients with no A and ≥2 BILAG-2004 B scores. These treatment group imbalances in baseline disease activity were not observed in any of the other subgroups of TULIP-1 or TULIP-2 (table 1, online supplemental table S3). Organ involvement at baseline is presented in online supplemental table S4.
SLE disease activity at baseline in TULIP-1 stratified by BICLA/SRI(4) response
In the TULIP-1 BICLA non-responder/SRI(4) responder subgroup, placebo and anifrolumab groups did not differ in the proportions of patients receiving oral glucocorticoids. However, mean daily glucocorticoid dose at baseline in the placebo group was lower than in the anifrolumab group, although SD were large (mean (SD), 9.5 (5.8) mg/day vs 11.6 (5.8) mg/day) (online supplemental table S5). The proportion of patients receiving glucocorticoids ≥10 mg/day who achieved taper to ≤7.5 mg/day was also lower in the placebo group than in the anifrolumab 300 mg group (8/15 (53.3%) vs 5/7 (71.4%)), although results should be interpreted with caution given the small group sizes (figure 2). These treatment group imbalances in glucocorticoid use were not observed in the concordant TULIP-1 subgroups.
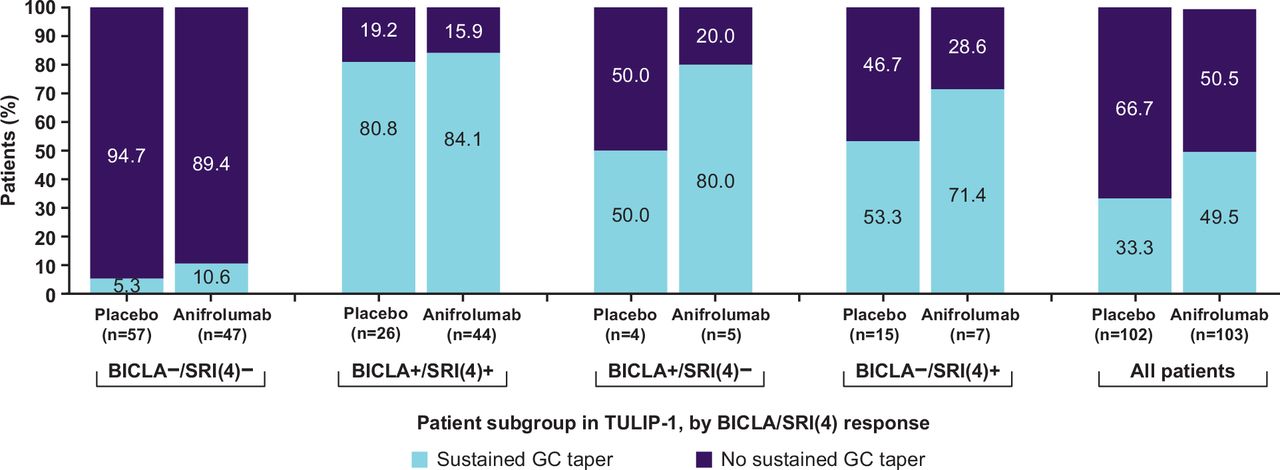
Proportion of patients with and without sustained taper of glucocorticoids to ≤7.5 mg/day from week 40 to week 52 among patients receiving ≥10 mg/day at baseline, stratified by BICLA/SRI(4) response in TULIP-1. All patients included in this analysis were receiving glucocorticoids (prednisone or equivalent) ≥10 mg/day at baseline. BICLA– and SRI(4)– refer to non-responders; BICLA+ and SRI(4)+ refer to responders. BICLA, British Isles Lupus Assessment Group-based Composite Lupus Assessment; GC, glucocorticoid; SRI(4), Systemic Lupus Erythematosus Responder Index of ≥4.
SRI(4) Response Characteristics in the TULIP-1 BICLA Non-responder/SRI(4) Responder Subgroup
Most placebo-treated patients in the TULIP-1 BICLA non-responder/SRI(4) responder subgroup attained an SRI(4) response as a result of their arthritis response (22/28, 78.6%) (figure 3A, table 2); 25.0% (7/28) of patients in the placebo group attained resolution only in the arthritis domain, whereas none of the 12 anifrolumab-treated patients had responses solely restricted to the arthritis domain (table 2). In the anifrolumab 300 mg group, domain improvements that led to SRI(4) responses showed more variation, with 6 (50%) patients attaining a SLEDAI-2K arthritis response. The proportion of patients with musculoskeletal responses at week 52 is also presented for the BICLA responder/SRI(4) responder, BICLA responder/SRI(4) non-responder, and BICLA non-responder/SRI(4) non-responder subgroups in online supplemental table S6; the proportion of patients with arthritis response was similar between treatment groups in the concordant subgroups. Only in the discordant BICLA non-responder/SRI(4) responder subgroup was an imbalance in arthritis responses seen, with more such patients in the placebo group.
Reasons for SRI(4) response at week 52 in TULIP-1 among BICLA non-responders/SRI(4) responders
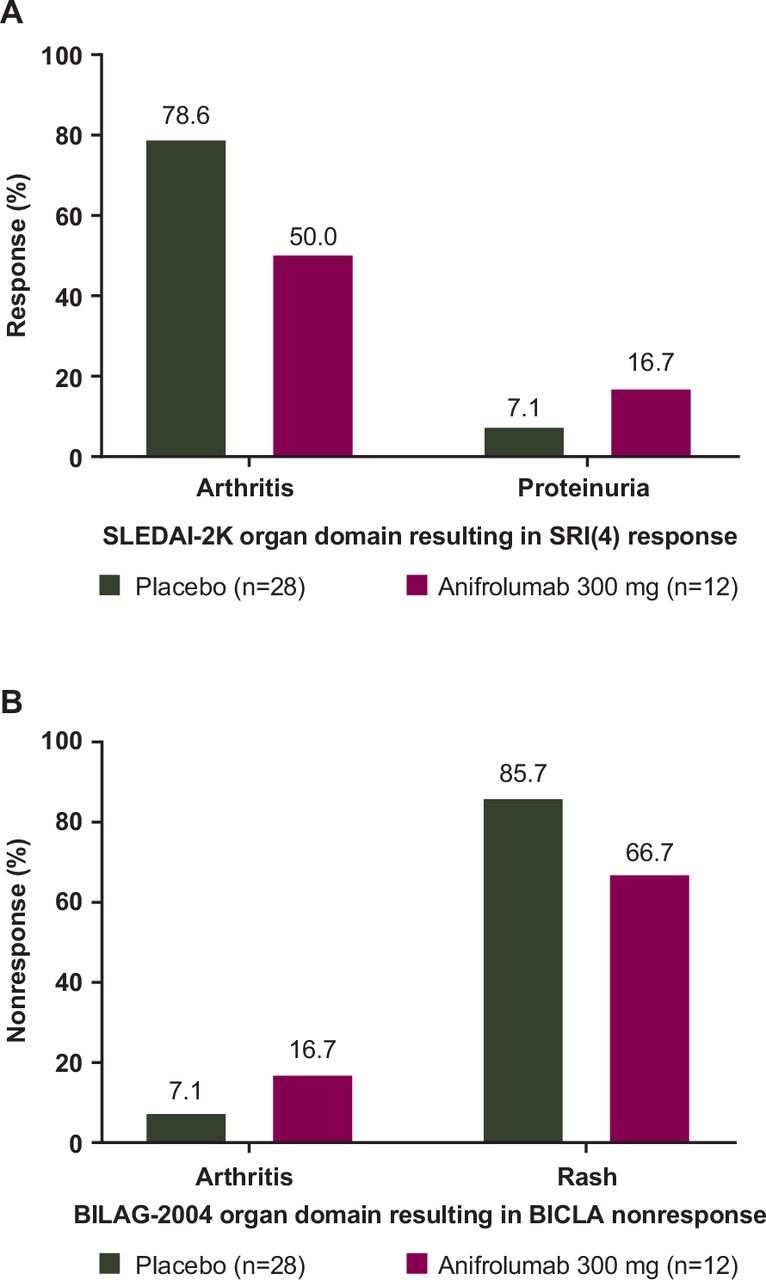
{kind=link}
{kind=link}
{kind=link}
Reasons for (A) SRI(4) response and (B) BICLA non-response at week 52 in TULIP-1 among BICLA non-responders/SRI(4) responders. BICLA– and SRI(4)– refer to non-responders; BICLA+ and SRI(4)+ refer to responders. BICLA, British Isles Lupus Assessment Group-based Composite Lupus Assessment; SLEDAI-2K, Systemic Lupus Erythematosus Disease Activity Index 2000; SRI(4), Systemic Lupus Erythematosus Responder Index of ≥4.
In light of the above findings, we determined the baseline joint counts of the 22 BICLA non-responders/SRI(4) responders in the placebo group who had SLEDAI-2K arthritis responses. Of these patients, 11 (50.0%) had <6 swollen and <6 tender joints at baseline, compared with 2/6 (33.3%) anifrolumab-treated patients in this subgroup (online supplemental figure S1). Baseline joint counts in the BICLA non-responders/SRI(4) responders in both the anifrolumab 300 mg and placebo groups were more varied.
Additionally, 12/22 placebo patients were receiving ≥10 mg/day glucocorticoid at baseline, 5 of whom (41.6%) were unable to taper glucocorticoids to ≤7.5 mg/day. In contrast, among the 6 anifrolumab-treated patients in this subgroup, 4 were receiving ≥10 mg/day glucocorticoid at baseline, all of whom were able to taper to ≤7.5 mg/day.
Reasons for BICLA Non-response in the TULIP-1 BICLA Non-responder/SRI(4) Responder Subgroup
In the TULIP-1 BICLA non-responder/SRI(4) responder subgroup, patients achieving a response on items resulting in a 4-point reduction in SLEDAI-2K also improved in the same organ domains on BILAG-2004. However, patients in this subgroup were BICLA non-responders because other moderate or severe organ involvement did not resolve. The most common reason for a BICLA non-response in this subgroup was a lack of improvement in BILAG-2004 rash in both the anifrolumab (8/12, 66.7%) and placebo groups (24/28, 85.7%) (figure 3B, online supplemental table S7). Overall, the combination of BICLA non-response due to rash and SRI(4) response due to arthritis occurred in 20 (71.4%) placebo patients and 5 (41.7%) anifrolumab-treated patients (table 3).
Overview of reasons for a BICLA non-response/SRI(4) response at week 52 in TULIP-1 among BICLA non-responders/SRI(4) responders
Discussion
Across the three trials in the anifrolumab clinical development programme for the treatment of patients with SLE, consistent BICLA and SRI(4) outcomes favouring anifrolumab were observed in TULIP-2 and MUSE, but not in TULIP-1.11–13 At an individual patient level, we identified a high level of concordance between the BICLA and SRI(4) composite endpoints across all trials. The proportion of patients in TULIP-1 who met the stringent ‘dual responder’ criteria was greater with anifrolumab than placebo and was similar to the effect size for ‘dual responders’ seen in TULIP-2, supporting a beneficial effect of anifrolumab on disease activity in patients with SLE. To our knowledge, this is the first time a ‘dual responder’ group has been defined in a clinical trial setting.
Discordant outcomes were observed in small proportions of patients in all three trials. In contrast to the TULIP-2 and MUSE trials, in which the proportions of discordant patients were similar in the anifrolumab and placebo treatment groups, BICLA non-responders/SRI(4) responders were more frequent in the TULIP-1 placebo group. This patient subgroup likely contributed to the lack of a significant SRI(4) treatment difference in TULIP-1.
In the TULIP-1 BICLA non-responder/SRI(4) responder subgroup, the primary reason for SRI(4) response was SLEDAI-2K resolution of arthritis (with a weight of 4 points). Arthritis improvement is sufficient to achieve individual SRI(4) responses, but alone is insufficient for a BICLA response unless all other baseline BILAG-2004 A and B organ activity improves.6 Baseline joint scores tended to be lower in the discordant placebo group than the anifrolumab group; therefore, SLEDAI-2K arthritis responses, and hence SRI(4) responses, may have been achieved more easily in the placebo group. In TULIP-1, residual clinical manifestations, which remained after arthritis improvement (predominantly rash), accounted for most of the patients classified as SRI(4) responders but BICLA non-responders.
In this same discordant TULIP-1 subgroup, smaller numbers of patients in the placebo vs the anifrolumab group achieved glucocorticoid taper. As such, the combination of less frequent glucocorticoid tapering and fewer active joints may have inflated the proportion of patients with an SRI(4) response in the placebo group. Fewer patients in the placebo group tapered glucocorticoids despite lower baseline joint involvement; this was surprising, since these patients may have been less likely to need high-dose glucocorticoids.
These variations in glucocorticoid tapering may reflect regional differences in standard therapy use. In TULIP-1, there was a baseline imbalance in patients enrolled from Eastern vs Western Europe between the anifrolumab and placebo groups. Regional differences in placebo group response rates have been previously identified, with higher SRI(4) response rates with standard therapy in patients from Eastern Europe and Central America than those in Western Europe or North America.24 In a previous international inception cohort study, there was significant between-centre variation in glucocorticoid use, even after adjustment for factors known to influence glucocorticoid dose.25 Our observations from TULIP-1 confirm that physician glucocorticoid prescribing behaviour varies25 which, if not accounted for, can contribute to an imbalance in trial outcomes. Inconsistent BICLA and SRI(4) outcomes should be expected given their different definitions and is consistent with previous findings in other trials. A previous phase 2 trial of epratuzumab reported similar disagreement between SRI(4) and BICLA, resulting in a higher placebo response rate using SRI(4).26 Despite similar SRI(4) and BICLA placebo response rates (33%) in a phase 2 trial of ustekinumab, SRI(4) response with ustekinumab was 62%, compared with a BICLA response of 35%.27 Two previous reviews comparing BICLA and SRI(4) concluded that, while both are viable tools for use as primary endpoints in SLE studies, differences in the trial populations and in study designs can impact the outcomes of each measure.15 28 Our findings add further evidence to support this conclusion.
This secondary analysis of TULIP-1 data provides important lessons for future SLE trial design. Variations in the number of active organ domains and joint counts at baseline, glucocorticoid prescribing/tapering practices and/or regions of trial recruitment may all increase the risk of discordant BICLA and SRI(4) outcomes. Baseline imbalances in these demographic and clinical factors potentially jeopardise the primary outcome; therefore, every effort should be made to ensure adequate balancing of these factors at study entry and during clinical trials. As regional differences in glucocorticoid tapering have been reported,25 additional sponsor-led training to normalise glucocorticoid tapering practice across centres may result in more standardised handling of background medications and more consistent placebo response rates in multicentre clinical trials.
In addition, setting minimum thresholds at enrolment for active joint counts, rash, oral ulcers and alopecia may improve the stringency of an endpoint such as SRI(4), which can be confounded by improvement in one or two highly-weighted organ domains in patients with low baseline disease activity.5 29 As SLEDAI-2K/SRI scoring may allow patients with lower joint counts to achieve the threshold for ‘response’, lupus trials may also benefit from the use of less subjective methods to assess lupus arthritis disease activity. This may require more refined clinical assessment of joints and/or imaging modalities such as MRI or ultrasound; however, imaging brings additional challenges of training and added expense, particularly in phase 3 trials.30 31
There were limitations in this study. This was a post hoc analysis, although of prospectively collected data. The numbers of patients in each treatment group in the discordant subgroups were relatively small, particularly for anifrolumab-treated patients, which prevents firm conclusions being drawn from comparisons between the anifrolumab and placebo groups. The complexity of trial outcomes and inclusion criteria may limit the extent to which these findings are generalisable to clinical practice. Furthermore, elements of the proposed explanation for the TULIP-1 SRI(4) discrepancy rely on circumstantial connections rather than a demonstrated causal relationship. However, analysis of future datasets may serve to validate these observations.
To conclude, in individual patient-level analyses, the majority of patients across the TULIP-1, TULIP-2 and MUSE trials of anifrolumab had concordant outcomes on BICLA and SRI(4). Using a stringent definition of response requiring dual BICLA and SRI(4) response, anifrolumab treatment was associated with efficacy compared with placebo in all three trials. A discordant BICLA non-responder/SRI(4) responder subgroup was identified in all three trials but this subgroup was larger in the TULIP-1 placebo group. Discordance was primarily driven by the sensitivity of SRI(4) to single organ (arthritis) improvement as the discordant placebo group was enriched for patients with lower baseline joint counts. The BICLA definition was less sensitive to this treatment group imbalance due to a requirement for at least partial improvement in all domains that were scored with moderate or severe BILAG-2004 scores at entry. Discordant placebo-treated patients showed regional recruitment variation, tended to have lower baseline disease activity and were less likely to taper glucocorticoids, providing additional reasons for higher placebo response rates. Given the emphasis placed on primary endpoint attainment in phase 3 trials by regulators, factors that jeopardise study outcomes need to be recognised and mitigated during trial design and execution. Confirmation of our observations in other trial cohorts may also suggest ways in which we can improve on current composite endpoints in a data-driven fashion. For now, we suggest that careful attention to baseline factors and maintaining uniformity in glucocorticoid tapering are essential in future SLE clinical trials to reduce the likelihood of discordant results and maximise the ability to detect efficacy signals.
Data availability statement
Data are available on reasonable request. Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
Ethics statements
Patient consent for publication
Ethics approval
The TULIP-1, TULIP-2 and MUSE trials were conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonisation Guidelines for Good Clinical Practice, and all patients provided written informed consent in accordance with local requirements. As this was a post hoc analysis of anonymized data, no ethics committee or institutional review board approvals were required—all such approvals were obtained in the original trials.
Acknowledgments
The authors would like to thank the investigators, research staff, healthcare providers, patients and caregivers who contributed to this study. The authors would like to thank Emma Witch of Audubon and Konstantina Psachoulia of AstraZeneca for their support and diligence for interpretation of patient-level analyses. Medical writing and editing assistance were provided by Rosie Butler, PhD, of JK Associates Inc., part of Fishawack Health. This support was funded by AstraZeneca. INB is a National Institute for Health Research (NIHR) Senior Investigator Emeritus and is funded by the NIHR Manchester Biomedical Research Centre. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
Correction notice This article has been corrected since it published Online First. A typographical error has been corrected in the abstract heading.
Contributors INB, RAF, EFM, MH and RT conceived and designed the study. EM collected the data. INB, RAF, EFM, SM, YT, KCK, JTM, PP, EM, CK, MA, MH and RT analysed and interpreted the data. INB, RAF, EFM, SM, YT, KCK, JTM, PP, EM, CK, MA, MH and RT were involved in development, review and final approval of the manuscript. All authors interpreted the data, critically reviewed the manuscript for important intellectual content, approved the final draft and agreed to its submission. INB accepts full responsibility for the finished work and/or the conduct of the study, had access to the data and controlled the decision to publish.
Funding The study was sponsored by AstraZeneca.
Competing interests INB has received grant/research support from Genzyme/Sanofi, GSK, Roche and UCB; received consulting fees from AstraZeneca, Eli Lilly, GSK, ILTOO, Merck Serono and UCB; and speaker/honoraria from AstraZeneca, GSK and UCB. INB is a National Institute for Health Research (NIHR) Senior Investigator Emeritus and is funded by the NIHR Manchester Biomedical Research Centre. RAF has received grant/research support and consulting fees from AstraZeneca. EFM received grant support from AstraZeneca, Bristol Myers Squibb, Janssen, Merck Serono and UCB; was a consultant for AstraZeneca, Eli Lilly, Janssen and Merck Serono; and was a speaker at a speaker bureau for AstraZeneca. SM has received grants and other support and has been a member of an advisory board for AstraZeneca. YT has received speaking fees and/or honoraria from AbbVie, Asahi Kasei, Astellas, Bristol Myers Squibb, Chugai, Daiichi-Sankyo, Eisai, Eli Lilly, Gilead, GSK, Janssen, Mitsubishi-Tanabe, Novartis, Pfizer, Sanofi and YL Biologics, and has received research grants from AbbVie, Chugai, Daiichi-Sankyo, Eisai, Mitsubishi-Tanabe, Takeda and UCB. KCK has received consulting fees from AbbVie, Amgen, AstraZeneca, Biogen, Chemocentryx, Eli Lilly, Equillium, Genentech/Roche, GSK, Janssen and Nektar; and has received grant/research support from BMS, Irdosia, Kirin, Pfizer, Resolve, Takeda and UCB. JTM has received grant/research support from BMS and GSK and has received consulting fees from AstraZeneca, AbbVie, Amgen, Aurinia, BMS, EMD Serono, GSK, Remegen, Janssen, Provention and UCB. PP, EM, CK, MA, MH and RT are employees of AstraZeneca.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.