Article Text

Abstract
Objectives To evaluate if baricitinib, a Janus kinase inhibitor, further enhances disease-modifying effects by uncoupling the link between disease activity and structural damage progression in patients with rheumatoid arthritis (RA) using two phase III randomised, double-blinded trials.
Methods In RA-BEAM, patients with established RA and inadequate response to methotrexate (MTX-IR) received placebo (PBO), baricitinib 4 mg or adalimumab 40 mg on background MTX. In RA-BEGIN, conventional synthetic disease-modifying antirheumatic drug (csDMARD)-naïve patients received MTX, baricitinib 4 mg or baricitinib 4 mg plus MTX. Using linear regression analyses, joint damage progression (assessed by change from baseline in van der Heijde modification of the Total Sharp Score) was compared between treatment groups for patients achieving certain disease activity states by the Clinical Disease Activity Index. Time-averaged postbaseline responses were used to week 24 (RA-BEAM) and week 52 (RA-BEGIN).
Results For MTX-IR patients, structural damage progression was reduced regardless of disease activity states in baricitinib-treated patients (p=0.6), whereas in PBO patients there was a clear dependence on disease activity states, being significantly lower in those who achieved remission/low disease activity (REM/LDA) compared with moderate/high disease activity (MDA/HDA) (p=0.02). Furthermore, the baricitinib MDA/HDA group had less damage progression than the PBO MDA/HDA group (p<0.001). For csDMARD-naïve patients, progression was lower in REM/LDA versus MDA/HDA within the MTX group (p<0.001). However, for baricitinib+MTX (p=0.5) or baricitinib monotherapy (p=0.07), progression was similar regardless of disease activity. In MDA/HDA groups, progression was lower with baricitinib+MTX (p<0.001) and numerically lower with baricitinib monotherapy (p=0.07) versus MTX. C reactive protein (≤5 mg/L and >5 mg/L) sensitivity analyses supported the primary findings.
Conclusions Baricitinib reduces structural damage progression versus PBO with background MTX and/or MTX, even in patients with MDA/HDA, showing a disease-modifying effect across all disease activity states.
- arthritis
- rheumatoid
- antirheumatic agents
- inflammation
- methotrexate
- outcome assessment
- health care
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
In patients with rheumatoid arthritis, tumour necrosis factor inhibitors, interleukin 6 inhibitors and rituximab have been shown to uncouple the link between disease activity and radiographic progression such that patients are protected from structural damage progression even if remission/low disease activity (REM/LDA) is not achieved.
What does this study add?
In two distinct populations of patients with rheumatoid arthritis (patients naïve to conventional synthetic disease-modifying antirheumatic drugs or with inadequate response to methotrexate (MTX)), either baricitinib alone and/or in combination with MTX enhanced disease-modifying properties by uncoupling the link between disease activity and structural damage progression, with the uncoupling being more evident for baricitinib in combination with MTX.
In the baricitinib groups, joint damage was controlled regardless of disease activity level, unlike in the control groups.
Patients with residual moderate or high disease activity who received baricitinib with background MTX or in combination with MTX had less structural damage progression than the control groups (MTX or placebo with background MTX).
Validation analysis showed a similar uncoupling of inflammation and structural damage progression when patients were stratified by high-sensitivity C reactive protein.
Key messages
How might this impact on clinical practice or future developments?
The uncoupling of disease activity and structural damage progression by baricitinib, a Janus kinase inhibitor, provides evidence which was not previously available for this mechanism of action.
Preservation of structural progression regardless of disease activity might ensure medium-term to long-term prevention of disability in patients who cannot achieve REM/LDA or require more time to reach this target.
This may inform treatment decisions in patient groups who have not, or have not yet, achieved sufficient clinical improvement.
Introduction
The high propensity to destroy cartilage and bone constitutes a major hallmark of rheumatoid arthritis (RA). While it appears that over the last two decades progression rates of joint damage have declined,1 patients with established disease entering clinical trials still have high baseline radiographic scores, implying aggressively damaging RA.2 Indeed, joint damage shows a significant positive association with both swollen joint counts and acute phase reactant levels.3–8 Joint swelling or synovitis characterises the local and acute phase reactants reflect the systemic inflammatory response, which usually go hand in hand, but joint swelling is more strongly associated with progression of joint damage than the acute phase response.4 9
Effective treatment with conventional synthetic disease-modifying antirheumatic drugs (csDMARDs) reduces the rate of structural progression and ultimately halts joint structural damage accrual once stringent remission is achieved and sustained afterwards.5 10–13 However, while joint damage progression is reduced on csDMARDs, structural deterioration still continues to occur in correlation with residual disease activity.
Several years ago, it was observed that the strong relationship between the extent of structural changes and inflammatory disease activity was blunted with infliximab use, a tumour necrosis factor α (TNF) inhibitor.14 In contrast to patients continuing methotrexate (MTX) treatment, who showed a linear progression of structural damage in relation to their disease activity state, patients who received infliximab plus MTX would not progress significantly, even if their disease activity remained moderate or high.15 Similar results were observed for other TNF inhibitors in later studies.16 17
The reason for the disruption of the tight link between disease activity and joint damage may be ultimately explained by a threshold effect: activation of osteoclasts, which induce bone erosions, requires higher TNF concentrations than induction of the inflammatory response. Therefore, when this cytokine is blocked to a level that still allows an unmitigated perpetuation of synovitis, this reduction may still suffice to not allow continued activation of osteoclasts, a major mediator of joint destruction.18
Interestingly, this interference with structural progression despite high disease activity is not confined to TNF inhibitors, but was also found for interleukin 6 (IL-6) inhibition and rituximab, usually combined with MTX.19 20 This suggests that biological (b)DMARDs reduce the inflammatory load of cytokines like TNF and IL-6 to a much larger extent than csDMARDs alone and thus downregulate the amplifying activity of these messenger molecules on osteoclastogenesis.21
Janus kinase (JAK) inhibitors (Jakinibs) belong to the most recent class of targeted synthetic (ts)DMARDs, which have shown at least similar efficacy as bDMARDs.22 JAKs are needed for signal transduction of various cytokine receptors, including those for IL-6 and interferons,23 but not TNF or most B cell receptors. Baricitinib, a Jakinib widely approved for treating RA, is a JAK1 and JAK2 inhibitor which has shown efficacy across RA subsets24–26; this efficacy includes structural damage inhibition. However, knowledge on structural damage reduction beyond the decrease in disease activity is limited. Indeed, when patients with poor prognostic factors were assessed for joint damage progression, another Jakinib, tofacitinib, failed to show a significant reduction compared with bDMARD.27
This question is the focus of research addressed in the present study.
Methods
Study design
In the 52-week RA-BEAM trial, adult patients with active RA (≥6/68 tender joints, ≥6/66 swollen joints and a high-sensitivity serum C reactive protein (hsCRP) ≥6 mg/L) and no previous bDMARD therapy were included.25 Patients had either ≥3 joint erosions or ≥1 joint erosions plus seropositivity for rheumatoid factor or anticitrullinated peptide antibodies (ACPA). Patients had an inadequate response to MTX (MTX-IR) and received background MTX throughout the study. Randomisation was 3:3:2 to placebo (PBO) (n=488), baricitinib 4 mg (n=487) and adalimumab 40 mg (n=330).
In RA-BEGIN, adult patients with active RA who had received no/limited treatment with csDMARDs (up to 3 weekly doses of MTX allowed) and no treatment with bDMARDs were included.24 Patients had ≥6/68 tender joints and ≥6/66 swollen joints and serum hsCRP level ≥3.6 mg/L and were seropositive for rheumatoid factor or ACPA. Randomisation was 4:3:4 to MTX (n=210), baricitinib 4 mg (n=159) and baricitinib 4 mg plus MTX (n=215) for 52 weeks.
Outcome measures
Joint damage progression was assessed using the van der Heijde modification of the Total Sharp Score (mTSS).28 29 Two independent readers, who were unaware of the chronological order, patient identity and treatment group, scored the radiographs and the mean score between the readers was used.
Disease activity was assessed by the Clinical Disease Activity Index (CDAI), stratified by the states of remission/low disease activity (REM/LDA; CDAI ≤10) and moderate/high disease activity (MDA/HDA; CDAI >10). Systemic inflammation was assessed by hsCRP stratified at ≤5 mg/L and >5 mg/L.
Analyses populations
In RA-BEAM, analyses were performed for endpoints at week 24 to allow comparison with the PBO group; at week 24 all patients randomised to PBO were switched to baricitinib 4 mg and therefore analyses at week 52 were not done. Analyses in RA-BEGIN were done for endpoints at week 52 because in this study patients who were randomised to MTX were followed up until week 52, unless they were rescued or discontinued from the study, and thus baricitinib could be appropriately compared with this control population in the longer term.
Only completers of the relevant study endpoint were included, excluding patients who switched treatment, were rescued or lost to follow-up before the time point defined for analysis. In RA-BEAM, 329, 427 and 273 patients in the PBO, baricitinib and adalimumab groups, respectively, were defined as completers. Patients with missing structural data and/or missing data on the covariates used in the models were also excluded from the analysis. Thus, 318, 407 and 262 patients, respectively, were included in the CDAI analysis in RA-BEAM. In RA-BEGIN, 142, 129 and 170 patients in the MTX, baricitinib monotherapy and baricitinib+MTX groups, respectively, were defined as completers. After excluding patients with missing data, 134, 125 and 166 patients, respectively, in these groups were included in the CDAI analysis.
Statistical analysis
Analyses of change from baseline in mTSS in RA-BEAM and RA-BEGIN have been done stratifying by treatment and by postbaseline CDAI response at week 24 and week 52, respectively. Adjusted means for the mTSS change from baseline were estimated using linear regression models adjusted by baseline CDAI. Adjusted means for change from baseline in mTSS responses are displayed as effects plots. The adjusted means were estimated from the multivariable models, with continuous covariates fixed at their mean values and categorical covariates fixed at their proportional distribution in the data.
As sensitivity analyses, similar analyses of change from baseline in mTSS were done stratifying by hsCRP. Adjusted means for the mTSS change from baseline, stratified by treatment and by postbaseline hsCRP response at week 24 in RA-BEAM and week 52 in RA-BEGIN, were estimated using linear regression models adjusted by baseline hsCRP.
Change from baseline in CDAI at week 24 in RA-BEAM and week 52 in RA-BEGIN in patients with postbaseline CDAI >10 was estimated using linear regression analyses (observed values). Similar analyses were undertaken for hsCRP in patients with elevated postbaseline hsCRP.
For both CDAI and hsCRP postbaseline responses, individual time point responses were averaged until the end of the period of analysis (week 24 or week 52) to define the response at the specific time point of analysis. This analytical process tends to diminish the potential influence of extreme values, especially for hsCRP, and also reduces the number of patients with missing data at individual endpoint visits. The definition of disease activity and hsCRP response using a time-averaged CDAI and hsCRP has been used previously.16
Results
Baseline demographics and patient characteristics
Baseline demographics and patient characteristics have been published previously.24 25 Summaries of these data for the analyses populations are presented in table 1 (CDAI response analyses) and table 2 (hsCRP sensitivity analyses). The characteristics of the different patient populations at baseline were mostly similar. However, reflective of the different patient populations in the two trials, patients in RA-BEGIN (early RA naïve to csDMARDs) had a shorter disease duration and had lower mTSS scores than patients in RA-BEAM (established RA and MTX-IR).
Demographic and baseline characteristics of patients in RA-BEAM (MTX-IR patients with established RA) and RA-BEGIN (csDMARD-naïve patients with early RA) stratified by averaged CDAI
Demographic and baseline characteristics of patients in RA-BEAM (MTX-IR patients with established RA) and RA-BEGIN (csDMARD-naïve patients with early RA) stratified by averaged hsCRP
Treatment response in relation to disease activity
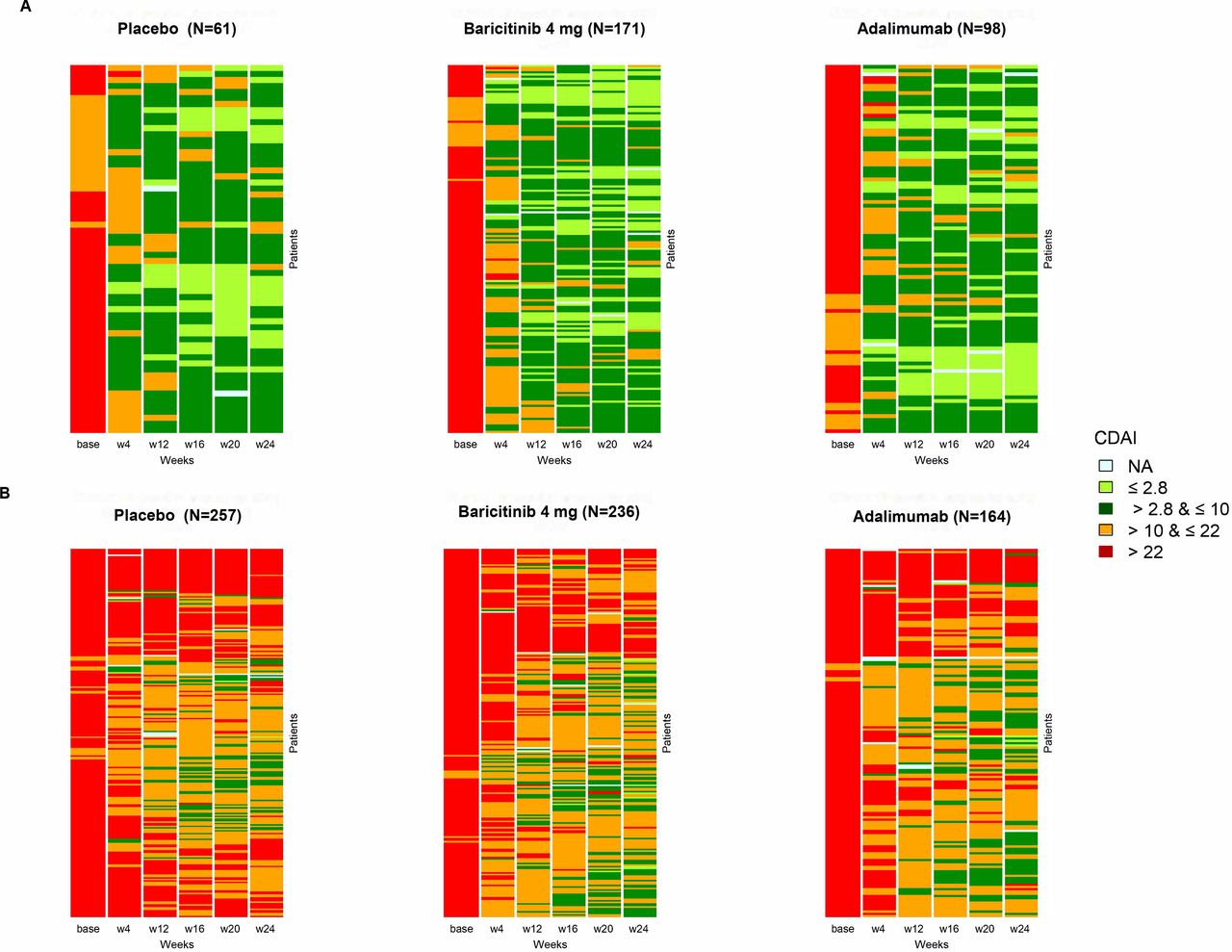
In RA-BEAM, 19.2% (n=61), 37.4% (n=98) and 42.0% (n=171) of patients in the analysed population who received PBO, adalimumab and baricitinib, respectively, achieved REM/LDA. Heatmap plots show individual CDAI longitudinal responses at all postbaseline visits for all patients included in the analyses (figure 1), differentiating REM/LDA and MDA/HDA CDAI responses at week 24.

Heatmaps showing individual CDAI responses to treatment in RA-BEAM (MTX-IR patients with established RA) in patients with (A) averaged CDAI ≤10 (remission/low disease activity) and (B) averaged CDAI >10 (moderate/high disease activity). Averaged CDAI responses calculated as the mean of postbaseline measurements at weeks 4, 12, 16, 20 and 24. CDAI, Clinical Disease Activity Index; MTX-IR, inadequate response to methotrexate; NA, not available; RA, rheumatoid arthritis; w, week.
In RA-BEGIN, at week 52, the proportion of patients in REM/LDA was 39.6% (n=53), 57.6% (n=72) and 62.7% (n=104), respectively, in the MTX, baricitinib monotherapy and baricitinib+MTX groups. Figure 2 displays the heatmap plots showing individual responses to treatment over time for patients stratified by CDAI responses at week 52.

Heatmaps showing individual CDAI responses to treatment in RA-BEGIN (csDMARD-naïve patients with early RA) in patients with (A) averaged CDAI ≤10 (remission/low disease activity) and (B) averaged CDAI >10 (medium/high disease activity). Averaged CDAI responses calculated as the mean of postbaseline measurements at weeks 4, 12, 16, 20, 24, 32, 40 and 52. CDAI, Clinical Disease Activity Index; csDMARDs, conventional synthetic disease-modifying antirheumatic drugs; MTX, methotrexate; NA, not available; RA, rheumatoid arthritis; w, week.
Structural damage progression in relation to disease activity in RA-BEAM (MTX-IR patients with established RA)
Among those achieving REM/LDA, patients who received PBO, adalimumab and baricitinib had adjusted means for mTSS change from baseline of 0.31, 0.15 and 0.24, respectively, compared with 0.84, 0.28 and 0.34, respectively, among patients in these groups with MDA/HDA (figure 3A). Thus, in the PBO group, patients achieving REM/LDA had less structural damage progression than patients with MDA/HDA (adjusted mean difference (95% CI): −0.53 (−0.98 to –0.09), p=0.02). In contrast, there was no significant difference in structural damage progression depending on disease activity states within patients receiving baricitinib (−0.09 (−0.41 to 0.22), p=0.6) or adalimumab (−0.13 (−0.53 to 0.26), p=0.5). Given that those with MDA/HDA did not have greater progression than those with REM/LDA, this reveals an uncoupling of disease activity and structural damage progression with baricitinib treatment.

Structural damage progression (adjusted mean for change from baseline mTSS) in relation to averaged CDAI in (A) RA-BEAM (MTX-IR patients with established RA) and (B) RA-BEGIN (csDMARD-naïve patients with early RA). Averaged CDAI responses in RA-BEAM calculated as the mean of postbaseline measurements at weeks 4, 12, 16, 20 and 24 and in RA-BEGIN as the mean of postbaseline measurements at weeks 4, 12, 16, 20, 24, 32, 40 and 52. REM/LDA classified as CDAI ≤10. Between treatment group difference, **p<0.001 versus placebo or MTX; within treatment group difference, †p<0.05 and ††p<0.001. CDAI, Clinical Disease Activity Index; csDMARDs, conventional synthetic disease-modifying antirheumatic drugs; LDA, low disease activity; mTSS, modified Total Sharp Score; MTX, methotrexate; MTX-IR, inadequate response to methotrexate; RA, rheumatoid arthritis; REM, remission.
Among patients with MDA/HDA, the adjusted mean difference (95% CI) for adalimumab compared with PBO was −0.56 (−0.87 to –0.25, p<0.001), in line with previous findings on the dissociative capacity of TNF inhibition on the link between disease activity and joint damage progression. The adjusted mean difference for baricitinib versus PBO was −0.50 (−0.78 to –0.23, p<0.001), indicating that, among those with MDA/HDA, there was significantly less structural damage progression in patients receiving baricitinib. This further suggests an uncoupling of disease activity and structural damage progression with baricitinib, which did not occur with PBO. There was no difference in structural damage progression between patients in the baricitinib and adalimumab groups (0.06 (−0.26 to 0.37), p=0.7).
Structural damage progression in relation to disease activity in RA-BEGIN (csDMARD-naïve patients with early RA)
Among patients who achieved REM/LDA, the adjusted means for mTSS change from baseline to week 52 were 0.43, 0.39 and 0.28 in the MTX, baricitinib monotherapy and baricitinib+MTX groups, respectively (figure 3B). Patients in the same treatment groups with MDA/HDA had progressed by 1.69, 1.05 and 0.50, respectively. Thus, patients receiving MTX in REM/LDA had significantly less structural damage progression compared with those with MDA/HDA (−1.26 (−1.95 to –0.57), p<0.001). In contrast, there were no significant differences in progression among patients who received baricitinib+MTX (−0.22 (−0.85 to 0.41), p=0.5) or those who received baricitinib in monotherapy (−0.65 (−1.36 to 0.06), p=0.07), although this difference was numerically larger for baricitinib monotherapy.
The magnitude of differences between the MTX (1.69), baricitinib (1.05) and baricitinib+MTX (0.50) groups among patients with MDA/HDA again reveals an effect of both baricitinib monotherapy and combination therapy on structural damage progression inhibition relative to MTX treatment. Compared with MTX, the adjusted mean difference for baricitinib monotherapy was −0.64 (−1.33 to 0.05, p=0.07) and was −1.19 (−1.85 to –0.53, p<0.001) for baricitinib+MTX.
These results indicate an uncoupling of disease activity and structural damage progression with combination therapy and a similar trend with baricitinib monotherapy, which did not occur with MTX monotherapy.
CDAI changes in patients with residual disease activity
In RA-BEAM, among all patients with a postbaseline averaged CDAI classed as HDA (n=229), more were in the PBO group than in the baricitinib and adalimumab groups (49.3%, 29.7% and 21%, respectively). The CDAI change from baseline to week 24 among patients with MDA/HDA was significantly greater in the baricitinib (−22.78) and adalimumab (−22.30) groups, compared with PBO (−17.26, both p<0.001; online supplemental table 1A). This indicates that even in patients who do not achieve REM/LDA, baricitinib conveys more clinical improvement and radiographic control than PBO, which is evidenced by the dissociation seen in this study (figure 3A).
Supplemental material
Similarly, among all patients with HDA (n=40) in RA-BEGIN, more were on de novo MTX (57.5%) than on baricitinib (15.0%) and baricitinib+MTX (27.5%). Interestingly, in contrast to RA-BEAM, among patients classed as having MDA/HDA in RA-BEGIN, the CDAI change from baseline on MTX (−24.8) was not significantly lower than on baricitinib monotherapy (−26.4) or combination therapy (−27.0) (both p>0.05; online supplemental table 1B). Nevertheless, structural progression was higher, suggesting that the capacity of baricitinib to dissociate the tight link between activity and damage goes beyond the mere association with change in disease activity or disease activity states (figure 3B).
Structural damage progression in relation to systemic inflammation
Regarding systemic inflammation, as reflected by hsCRP levels, 80.3% (n=257), 40.4% (n=107) and 36.3% (n=149) of patients receiving PBO, adalimumab and baricitinib, respectively, had a postbaseline averaged hsCRP >5 mg/L up to 24 weeks in RA-BEAM.
The impact of the systemic inflammatory response on structural damage progression within each treatment group differed (figure 4A). Among patients receiving PBO, those with hsCRP >5 mg/L had significantly greater structural damage progression compared with those with hsCRP ≤5 mg/L (0.48 (0.04 to 0.91), p=0.03). However, there were no significant differences among patients receiving baricitinib (0.18 (−0.14 to 0.50), p=0.3) or adalimumab (0.12 (−0.26 to 0.51), p=0.5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Structural damage progression (adjusted mean for change from baseline mTSS) in relation to averaged hsCRP in (A) RA-BEAM (MTX-IR patients with established RA) and (B) RA-BEGIN (csDMARD-naïve patients with early RA). Averaged hsCRP in RA-BEAM calculated as the mean of postbaseline measurements at weeks 4, 12, 16, 20 and 24 and in RA-BEGIN as the mean of postbaseline measurements at weeks 4, 12, 16, 20, 24, 32, 40 and 52. Between treatment group differences, *p<0.05 and **p<0.001 versus placebo or MTX; within treatment group differences, †p<0.05. csDMARDs, conventional synthetic disease-modifying antirheumatic drugs; hsCRP, high-sensitivity C reactive protein; mTSS, modified Total Sharp Score; MTX, methotrexate; MTX-IR, inadequate response to methotrexate; RA, rheumatoid arthritis.
Across treatment groups, among patients with hsCRP >5 mg/L, structural damage progression was lower in those receiving baricitinib compared with PBO (−0.45 (−0.77 to –0.13), p=0.006). Structural damage progression was also lower in patients who received adalimumab versus PBO (−0.55 (−0.90 to –0.20), p<0.01). As in the clinical assessment analyses, there was no essential difference between the baricitinib and adalimumab groups (0.10 (−0.29 to 0.49), p=0.6).
These results indicate that structural damage progression was uncoupled from inflammation in patients receiving baricitinib and show that, even with high systemic inflammation, structural damage progression was inhibited by baricitinib.
In RA-BEGIN, at week 52, 59.1% (n=81), 42.5% (n=54) and 28.1% (n=47) of patients in the MTX, baricitinib and baricitinib+MTX groups, respectively, had hsCRP >5 mg/L. These patients had progressed by 1.51, 1.20 and −0.15, respectively (figure 4B). Structural damage progression was significantly lower in the baricitinib+MTX group compared with the MTX group (−1.66 (−2.36 to –0.95), p<0.001). Damage progression in patients who received baricitinib monotherapy was also numerically lower compared with those who received MTX, but this was not significant (−0.31 (−0.97 to 0.36), p=0.4).
Within treatment groups stratified by acute phase response, structural damage progression was significantly greater in patients with hsCRP >5 mg/L in the MTX (0.73 (0.07 to 1.40), p=0.03) and baricitinib monotherapy (0.89 (0.21 to 1.57), p=0.01) groups, but did not significantly differ in the baricitinib+MTX group (−0.67 (−1.35 to 0.02), p=0.06).
hsCRP changes in patients with residual inflammation
In RA-BEAM, in patients with averaged hsCRP >5 mg/L, the change from baseline in hsCRP was significantly greater in the baricitinib group (−6.61, p<0.001), but not in the adalimumab group (−0.75, p=0.64), compared with PBO (online supplemental table 2A). This might be in line with the direct effect of baricitinib on IL-6 signalling and thus on C reactive protein (CRP). However, the fact that the change of hsCRP on adalimumab was not different from that in PBO again reveals that the dissociation is largely independent of changes of acute phase reactant levels in non-responders, confirmed by the similar inhibitory effect of baricitinib on progression of damage as that of adalimumab (figure 4A). This is further exemplified in RA-BEGIN. The difference in change from baseline in participants with averaged hsCRP >5 mg/L was −1.71 (p=0.35) for baricitinib and −3.94 (p=0.045) for baricitinib+MTX, compared with MTX alone (online supplemental table 2B), although MTX did not inhibit structural damage progression to the same extent as baricitinib, particularly when in combination with MTX (figure 4B).
Discussion
The research presented here originates from observations that in csDMARD-naïve patients with RA, MTX alone might not halt the progression of joint damage if there is ongoing residual disease activity. Early in the disease, as part of the window of opportunity, this is not acceptable, and patients should optimise treatments; thus far, tsDMARDs and bDMARDs are approved and recommended options. Later in the disease, in MTX-csDMARD insufficient responders who receive PBO, damage accrual is high, in line with their continued active established disease; in patients who receive either tsDMARDs or bDMARDs (with/without background MTX, but more so with combination therapy), damage could be halted or dramatically reduced even if they continue to have MDA/HDA.14–17 19 20 These analyses confirmed this hypothesis for baricitinib.
The results presented reveal that baricitinib, with/without de novo MTX or with background MTX, enhances disease-modifying effects by blunting the tight link that is usually seen between disease activity and progression of joint damage in csDMARD-naïve and MTX-IR patients, being more evident when baricitinib is combined with MTX. Thus, baricitinib and baricitinib+MTX exert efficacy in structural terms even in patients who remain in MDA/HDA on this treatment, being significant for baricitinib+MTX. This was also evident when changes from baseline in disease activity were examined in those with residually active disease.
These data are very robust on several grounds. First, they are independent of disease duration and prior treatment. They pertain to patients with early disease who are MTX-naïve, as exemplified in the RA-BEGIN trial analyses, and to patients with established disease who had an insufficient response to MTX, as evaluated in RA-BEAM. Second, the analyses are consistent and confirmatory irrespective of the type of inflammatory marker used. When subgroups for the definition of disease activity are formed according to clinical assessment (CDAI), which is primarily driven by joint counts and thus local inflammation, damage progression is not larger in higher versus lower disease activity states on baricitinib+MTX in contrast to control (MTX in RA-BEGIN and PBO with background MTX in RA-BEAM). These data are confirmed when employing CRP, a systemic inflammatory marker induced by proinflammatory cytokines in the liver, to distinguish patients with higher and lower disease activity.
In RA-BEGIN baricitinib monotherapy was also studied. While there was a trend towards better structural efficacy in patients with higher disease activity states also with baricitinib monotherapy compared with MTX monotherapy, this did not reach statistical significance. It should be considered that in early disease if uptitration of MTX does not control inflammation, structural progression would be higher than with use of a bDMARD or Jakinib, such as baricitinib, as indicated by mTSS progression over time. However, other studies of bDMARDs and tsDMARDs on csDMARD/MTX-naïve patients with early RA have not compared monotherapy, MTX combination therapy and MTX uptitration.
One of the strengths of the present study is the use of time-averaged disease activity, for both CDAI and CRP, so that the effects of extreme values and missing data are mitigated. Interestingly, when following disease activity in individual patients over time, in patients in whom REM/LDA is achieved at endpoint, a drop in activity is already discernible within 4–12 weeks, in line with the treat-to-target recommendations2 and independent of whether patients have established or early RA. In contrast, those who remain in MDA/HDA never achieve any better status.30 The visualisation as ‘heatmaps’ allowed us to show these data for individual patients very clearly. Another strength is the use of the CDAI for clinical disease activity assessment rather than a single measure. Composite scores capture RA better than individual variables.31 32 Because the CDAI does not include an acute phase reactant,7 we could validate the clinical findings by using a serological marker.
Our study has some limitations. First, we used different time points for analysis of the early csDMARD-naïve and established MTX-IR RA populations. While the ideal time frame for comparative assessment of radiographic progression is 1 year, as done in the RA-BEGIN analysis, assessments of RA-BEAM data were restricted to the 24-week time point. However, it was more important to have a valid active comparator, and in RA-BEAM all patients in the PBO group received baricitinib after at most 6 months; analyses at 1 year would then have confounded the value of the data. Interestingly though, significant differences between the groups could already be seen at 6 months, which may even increase the importance of the data. Another limitation is our focus on baricitinib and therefore we cannot be sure that our findings can be translated to other Jakinibs. While it is likely this will be the case, there might be some differences based on the different selectivity of the compounds.
In conclusion, the Jakinib baricitinib has shown to have significant inhibitory effects on the progression of structural joint damage even in patients who continue to have MDA/HDA states. This quality has hitherto been described only for bDMARDs and has important clinical value. Adherence to treat-to-target principles calls for a rapid change of treatment with insufficient improvement. However, when a patient improves clinically on baricitinib but not yet to the desired extent,33 the decision to change treatment could be delayed for a short while in accordance with the patient because joint damage progression and thus irreversible disability need not be feared.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants. The studies were approved by each centre's institutional review board or ethics committee. Participants gave informed consent to participate in the study before taking part.
Acknowledgments
The authors thank Andrea Hemmingway, PhD (Eli Lilly and Company) for writing and editorial assistance, and Liliana Zaremba-Pechmann, PhD (HaaPACS) for validation of statistical analyses.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor David S Pisetsky
Contributors PLR, IdlT, and JSS were involved in conception and design of the work. PLR designed and completed the statistical analyses. All authors contributed to data interpretation, critically reviewed the manuscript, and approved the final version for submission. PLR acts as guarantor for this work.
Funding The RA-BEGIN and RA-BEAM studies were supported by Eli Lilly and Company and Incyte Corporation. Medical writing and editorial support for this manuscript were funded by Eli Lilly and Company.
Competing interests PLR developed this work while an employee of Eli Lilly and Company and is currently an employee of Novartis. IdlT and EH are employees of Eli Lilly and Company. DA reports grants/contacts from AbbVie, Amgen, Eli Lilly and Company, Novartis, Roche, SoBi and Sanofi; and consulting fees from AbbVie, Amgen, Eli Lilly and Company, Merck, Novartis, Pfizer, Roche and Sandoz. JSS reports grants/contracts from AbbVie, AstraZeneca, Eli Lilly and Company and Novartis; consulting fees from AbbVie, Amgen, AstraZeneca, Astro, Bristol Myers Squibb, Celgene, Celltrion, Chugai, Eli Lilly and Company, Gilead, ILTOO, Janssen, Merck Sharp & Dohme, Novartis-Sandoz, Pfizer, Roche, Samsung, Sanofi and UCB; payment/honoraria from AbbVie, Amgen, Bristol Myers Squibb, Celgene, Chugai, Eli Lilly and Company, Janssen, Merck Sharp & Dohme, Novartis-Sandoz, Pfizer, Roche, Samsung and UCB; participation on a data safety monitoring/advisory board for AstraZeneca; a leadership/fiduciary role with European League Against Rheumatism; and other interests as an editorial board member of Annals of the Rheumatic Diseases and textbook editor.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.