Article Text

Abstract
Objectives S100A9, an alarmin that can form calprotectin (CP) heterodimers with S100A8, is mainly produced by keratinocytes and innate immune cells. The contribution of keratinocyte-derived S100A9 to psoriasis (Ps) and psoriatic arthritis (PsA) was evaluated using mouse models, and the potential usefulness of S100A9 as a Ps/PsA biomarker was assessed in patient samples.
Methods Conditional S100A9 mice were crossed with DKO* mice, an established psoriasis-like mouse model based on inducible epidermal deletion of c-Jun and JunB to achieve additional epidermal deletion of S100A9 (TKO* mice). Psoriatic skin and joint disease were evaluated in DKO* and TKO* by histology, microCT, RNA and proteomic analyses. Furthermore, S100A9 expression was analysed in skin, serum and synovial fluid samples of patients with Ps and PsA.
Results Compared with DKO* littermates, TKO* mice displayed enhanced skin disease severity, PsA incidence and neutrophil infiltration. Altered epidermal expression of selective pro-inflammatory genes and pathways, increased epidermal phosphorylation of STAT3 and higher circulating TNFα were observed in TKO* mice. In humans, synovial S100A9 levels were higher than the respective serum levels. Importantly, patients with PsA had significantly higher serum concentrations of S100A9, CP, VEGF, IL-6 and TNFα compared with patients with only Ps, but only S100A9 and CP could efficiently discriminate healthy individuals, patients with Ps and patients with PsA.
Conclusions Keratinocyte-derived S100A9 plays a regulatory role in psoriatic skin and joint disease. In humans, S100A9/CP is a promising marker that could help in identifying patients with Ps at risk of developing PsA.
- psoriatic arthritis
- cytokines
- inflammation
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution 4.0 Unported (CC BY 4.0) license, which permits others to copy, redistribute, remix, transform and build upon this work for any purpose, provided the original work is properly cited, a link to the licence is given, and indication of whether changes were made. See: https://creativecommons.org/licenses/by/4.0/.
Statistics from Altmetric.com
WHAT IS ALREADY KNOWN ON THIS TOPIC
S100A9 is an alarmin that forms calprotectin (CP) heterodimers with S100A8, which is highly expressed in inflammatory skin diseases, such as psoriasis (Ps), but how S100A9-expressing keratinocytes contribute to Ps, and whether these affect psoriatic arthritis (PsA), is still unknown.
WHAT THIS STUDY ADDS
First-time investigation of the role of epidermal-derived S100A9 in vivo using genetically modified mouse models, providing a better appreciation of S100A9 complexes in cells, tissues and the whole organism.
Demonstration that epidermal-specific inactivation of S100A9 increases Ps-like severity and PsA incidence, suggesting that keratinocyte-derived S100A9 is protective in chronic skin and joint inflammation.
Side-by-side evaluation of circulating S100A8, S100A9 and CP (S100A8/S100A9) in patients with Ps and PsA.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study indicates that targeting S100 proteins might help treat skin and joint inflammation. Testing whether circulating S100A9/CP could help in identifying patients with Ps at risk of developing PsA in larger and appropriately designed studies is crucial to improve disease outcomes, prevent disability and reduce healthcare and societal costs.
Introduction
Psoriatic disease develops from a complex cross-talk between proliferating keratinocytes and infiltrating immune cells that leads to secretion of various cytokines and chemokines including IL-17, IL-21, IL-22, IL-6, IL-1β, TNFα and CXCL1/3/5, which initiate a pro-inflammatory systemic response.1 In 30% to 40% of patients with psoriasis (Ps), the disease is complicated by psoriatic arthritis (PsA).2 In 75% of these cases, skin involvement precedes joint inflammation, usually with a gap of 5–10 years between the appearance of psoriatic plaques and the first signs of arthritis.2 3 Early PsA diagnosis and intervention are critical to avoid joint damage that leads to impaired physical function, fatigue, depression and poor quality of life.4–6 The most common therapies for psoriatic disease target several cytokines such as TNFα, IL-12/23- and IL-17A, with usually better efficacy on the skin than on the joint manifestations.7 Little is known about the mediators involved in PsA development. Therefore, there is unmet need for novel diagnostic markers unique to PsA and for identifying the molecular determinants of skin–joint crosstalk.6 8
Genetically engineered mouse models (GEMMs) with inducible epidermal deletion of c-Jun and JunB (DKO*) revealed the function and/or therapeutic potential of distinct Ps/PsA mediators, such as TNFR signalling,9 S100A9,10 VEGF11 and thymic stromal lymphopoietin12 as well as microRNAs13 and amygdalin analogues.14 These DKO* mice present a psoriasis-like inflammatory skin disease with articular changes strongly resembling PsA.9 Thus, skin-specific genetic interventions can trigger PsA-like disease.
S100A8 and S100A9 are two calcium binding proteins upregulated in inflammatory conditions that form homodimers or S100A8/A9 heterodimers termed calprotectin (CP).10 15–17 During skin inflammation, the main S100A9 expressing cells are keratinocytes, neutrophils and macrophages.18 19 S100A9 accounts for up to 40% of cytosolic proteins in neutrophils,20 acts intracellularly by modulating the cytoskeleton, and extracellularly by recruiting other immune cells to inflammation sites and upregulating pro-inflammatory cytokines.19 21 CP has important anti-microbial properties22 and faecal CP is a validated clinical biomarker for gut inflammation.23
We previously reported that global S100A9 inactivation alleviated skin and joint inflammation in the DKO* mouse model.10 How S100A9-expressing keratinocytes contribute to Ps, and whether these affect PsA development, is still unknown. Here, we crossed a newly generated S100A9 floxed allele into the DKO* model to inactivate S100A9 only in epidermal cells. We tested whether and how epidermal expression of S100A9 influences Ps-like and PsA-like disease and whether circulating S100A9, S100A8 and CP could be used as markers for PsA.
Results
Role for epidermal S100A9 in psoriasis-like disease
DKO* mice were crossed with S100A9 floxed mice to generate a new GEMM with inducible triple epidermal deletion of c-Jun, JunB and S100A9 (TKO*). Deletion of the floxed alleles in keratin 5–positive basal epidermal cells of adult DKO* and TKO* mice was achieved by intraperitoneal tamoxifen injections (online supplemental figure S1A). Ps-like and PsA-like disease developed in both DKO* and TKO* mice within 2 weeks after the last injection (online supplemental figures S1B and S2A). S100A9 deletion in TKO* was first assessed by immunofluorescence (figure 1A). Ear sections from DKO*-S100A9−/− mice10 were used to confirm antibody specificity and JunB immunofluorescence included for comparison. As previously reported,10 12 JunB expression in DKO* epidermis is patchy, while S100A9 is readily detectable in all lesional keratinocytes and in dermis-infiltrating immune cells. In TKO* mice, S100A9 expression appeared similar to that of JunB with a mosaic staining pattern, while dermis-infiltrating cells still expressed S100A9 (figure 1A). In TKO* mice, S100A9 was expressed in less than 18% of lesional epidermis in ears (figure 1B).
Supplemental material

Characterisation of psoriasis-like mouse model with inducible epidermal deletion of S100A9. (A) Immunofluorescence images of the ear skin of mice with inducible dual epidermal deletion of c-Jun and junB (DKO*), inducible triple epidermal deletion of c-Jun, JunB and S100A9 (TKO*) and DKO* mice with total deletion of S100A9 (DKO*-S100A9−/−) 23 days after first tamoxifen (TAM) injection (red: S100A9, green: JunB, scale bar=50 µm). (B) Quantification of S100A9-positive epidermal cells in the ear of DKO*, TKO* and DKO*-S100A9−/− mice (n=4–6). (C) Skin disease severity scoring in DKO* and TKO* mice. (D) Weight of control wild-type (WT), DKO* and TKO* with moderate/severe skin phenotype 23 days after first TAM injection (n=18–26). (E–J) S100A9 (E), S100A8 (F), calprotectin (G), IL-17A (H), IL-6 (I) and TNFα (J) concentrations in the sera of WT, DKO* and TKO* mice with moderate-severe psoriasis-like phenotype (n=4–13).

Psoriatic-arthritic-like (PsA) phenotype in mice with severe psoriasis-like disease. (A) Prevalence of psoriatic arthritis (PsA) in mice with inducible dual epidermal deletion of c-Jun and junB (DKO*) and triple epidermal deletion of c-Jun, JunB and S100A9 (TKO*) with severe psoriasis-like disease (n=20 per group). (B) Toluidine blue staining of the distal interphalangeal (DIP) joint of control wild-type (WT), DKO* and TKO* mice with PsA (scale bar=200 µm). (C) Quantification of toluidine blue staining intensity of articular cartilage in WT, DKO* and TKO* mice (WT n=4; DKO* n=8; TKO* n=6; each point represents the median of several joints measured per sample). (D) H&E-stained histological images showing psoriatic nail involvement with changes in the nail plate (P), nail matrix (M) and nail bed (B) of DKO* and TKO* mice and quantification of nail lesions (scale bar=100 µm). (E) H&E histological images of the distal phalanx (DP) showing enthesitis with high immune infiltration in the areas around the bone (scale bar=100 µm). (F) H&E histological images showing osteitis of the bone marrow (BM) of the distal phalanx (DP) in DKO* and TKO* mice (BM=bone marrow) with quantification of per cent area of bone marrow covered by inflammation (scale bar=100 µm).
Absence of epidermal S100A9 leads to more severe psoriatic skin disease
A macroscopic Ps-like classification was established as a function of ear inflammation/plaques and ventral skin inflammation (online supplemental figure S1B). In DKO* mice, the extent of weight loss and serum IL-17A and S100A9 levels correlated with skin disease severity (online supplemental figure S1C-E). Importantly, the skin of TKO* mice was overall more severely affected than DKO* (figure 1C), suggesting that keratinocyte-derived S100A9 inhibits severe skin inflammation.
While weight loss was similar between the two groups (figure 1D), DKO* and TKO* mice with moderate to severe skin phenotype had higher circulating S100A8 and S100A9 and similar CP (S100A8/A9), when compared with wild-type littermates (figure 1E–G). However, there was no difference between the two groups indicating that keratinocytes are not the major contributor to serum S100A9-containing dimers (figure 1E–G). Psoriasis-associated cytokines IL-17A, IL-6 and TNFα were also elevated in DKO* and TKO* sera compared with controls, but only TNFα was higher in TKO* (figure 1H–J). These data suggest that simultaneous inactivation of c-Jun, JunB and S100A9 in epithelial cells leads to an increase in Ps-like skin disease severity.
Absence of epidermal S100A9 leads to more severe psoriatic arthritis
Next, DKO* and TKO* mice with severe skin phenotype were macroscopically scored for signs of nail and entheseal involvement resembling PsA. While macroscopic swelling in the distal interphalangeal (DIP) joints appeared comparable between DKO* and TKO* (online supplemental figure S2A), a significant increase in PsA prevalence was observed in TKO* mice (figure 2A). Cartilage degradation was assessed by toluidine blue staining in the third DIP joint of the right hind limb. Compared with controls, decreased proteoglycan was observed in the articular region, but the extent of proteoglycan loss was comparable between DKO* and TKO* (figure 2B,C). Histological evaluation revealed extensive nail disease (figure 2D), enthesitis in the distal phalanx (figure 2E) and bone marrow osteitis in the distal phalanx (figure 2F) in both DKO* and TKO* mice. As PsA is associated with bone loss, we next quantified bone in the hind limbs using radiography and micro-CT (online supplemental figure S2B–I). Bone loss was apparent in both DKO* and TKO* mice, but not in controls, and was consistent with increased in serum IL-17.24 However, bone loss was not different in DKO* and TKO* mice (online supplemental figure S2B–I). Overall, these data indicate that keratinocyte-derived S100A9 decreases the incidence of PsA in the context of severe Ps-like skin disease, but does not affect the severity of PsA once it develops.
Epidermal S100A9 modulates neutrophil recruitment to inflammatory sites
Immunofluorescence co-staining of S100A9 and Ly6B, a surface marker expressed by neutrophils, inflammatory monocytes and some activated macrophages, was performed on whole ear sections from WT, DKO* and TKO* mice (figure 3A). Computer-assisted quantification was performed after digital removal of autofluorescent cartilage areas and neutrophil-rich, but difficult to quantify Munro micro-abscesses (online supplemental figure S3A). Ly6B+ cells were increased in DKO* skin sections compared with wild-type littermates and further significantly increased in TKO* skin sections (figure 3B). Around 50% of Ly6B+ cells also expressed S100A9 and a similar difference of 2–3 folds in absolute numbers was observed between DKO* and TKO* when considering Ly6B/S100A9 double-positive cells (figure 3C). Abundant S100A9+Ly6B+ myeloid cells were also observed in the inflamed joints of DKO* and TKO* mice (figure 3D–F). While synovial neutrophils were similarly increased in DKO* and TKO*, S100A9-positive neutrophils were more abundant in TKO* than DKO* mice (figure 3E–F). Interestingly, both Ly6B-positive and Ly6B/S100A9-double positive cells were increased in the bone marrow of TKO* relative to DKO* (online supplemental figure S3B–D), pointing to a possible contribution of these cells to increased PsA incidence in TKO*. These data suggest that S100A9-expressing neutrophils infiltrate skin and joints in DKO* and TKO* mice and that increased neutrophils might potentiate skin and joint disease in TKO* mice.

Neutrophil infiltration in mice with severe psoriasis-like disease. (A) S100A9 (red) and Ly6B (neutrophils; green) immunostaining of lesional ears in mice with inducible dual epidermal deletion of c-Jun and junB (DKO*) and triple epidermal deletion of c-Jun, JunB and S100A9 (TKO*) (dotted lines represent the basal membrane, yellow arrows point to Ly6B/S100A9-double positive cells, scale bar=20 µm). (B–C) Confocal microscopy–based quantification of absolute number of neutrophils (Ly6B-positive) (B) and S100A9-positive neutrophils (C) in the whole ear sections of DKO* and TKO* mice and wild-type (WT) littermates (n=4–5 mice). (D) S100A9 (red) and Ly6B (green) immunostaining of lesional psoriatic arthritis (PsA)–like paws (scale bar=200 µm). Yellow arrows point to infiltrating cells. (E–F) Confocal microscopy–based quantification of neutrophil (Ly6B-positive) (E) and S100A9-positive neutrophil (F) in the distal interphalangeal joints of DKO* and TKO* mice and WT littermates (average of 3–5 regions per paw, n=4 mice).
Epidermal S100A9 affects neutrophil-related proteins and pathways in skin
Mass spectrometry–based proteomics of whole ear lysates was next performed and volcano plots identified statistically significant proteins upregulated and downregulated in TKO* and DKO* mice, when compared with each other (figure 4A) or to control mice (online supplemental figure S4A,B). Venn diagrams identifiedproteins uniquely upregulated in each comparison as well as shared proteins (figure 4B). Enrichment analyses identified the most relevant pathways in each comparison and peptides statistically significant in at least one comparison were displayed in a heatmap, grouped by Gene Ontology terms and connected to their respective biological processes in an Alluvial plot (figure 4C). All these analyses revealed a largely predominant neutrophil activation signature in TKO* mice, consistent with our histological observations. TNF and Wnt signalling, which are involved in inflammation and aberrant bone formation, respectively, were also enhanced in TKO* mice, while other biological processes such as cellular response to IL-12 were similarly enriched in DKO* and TKO*. A connectivity network further confirmed the relevance of neutrophil granulation, innate immunity and TNF-mediated signalling in TKO* skin proteome with 76, 22 and 16 nodes, respectively (online supplemental figure S4C).

Proteomic analyses in whole ear extracts of DKO* and TKO* mice. (A) Volcano plot showing upregulated (red) and downregulated (blue) proteins (n=3–5 per condition; p<0.05); proteins in grey are below statistical significance. (B) Venn diagram depicting statistically significant upregulated proteins in (I) mice with inducible dual epidermal deletion of c-Jun and junB (DKO*) compared towild-type (WT) controls, (II) mice with triple epidermal deletion of c-Jun, JunB and S100A9 (TKO*) compared toWT mice and (III) TKO* compared to DKO* mice (n=3–5 per condition; p<0.05). (C) Heat map of significantly upregulated proteins (left) and Alluvial plot (right) of enriched biological processes associated to each protein.
Epidermal S100A9 modulates cytokine and chemokine expression during skin inflammation
Ear epidermis was isolated from DKO* and TKO* mice and littermate controls, dissociated and subjected to FACS analysis and sorting (online supplemental figure S5A). A similar increase in CD45+ immune cells was observed in DKO* and TKO* epidermal samples (online supplemental figure S5B,C) when compared with controls. Ly6G/CD11b double-positive neutrophils were higher in TKO* than DKO*, although not reaching statistical significance (online supplemental figure S5B-D). This finding indicates that when including Munro’s microabscesses, the overall number of immune cells and neutrophils in the epidermal area is comparable between the two genotypes. FACS-sorted neutrophils (figure 5A–F) and keratinocytes (figure 5G–L) were next analysed for cytokine and chemokine expression. A similar increase in s100a9 and s100a8 mRNA was observed in neutrophils isolated from DKO* and TKO* mice, compared with controls (figure 5A,B). il-1b, il-6 and tnf-a mRNA were similarly increased in neutrophils isolated from DKO* and TKO* mice, although most changes were not statistically significant (figure 5C–E). Interestingly, mRNA expression of osm, encoding the IL-6 family cytokine Oncostatin-M that induces psoriasis-like lesions in mice,25 appeared increased in neutrophils isolated from TKO* mice (figure 5F). Overall, neutrophils isolated from DKO* and TKO* epidermis display a similar pro-inflammatory mRNA expression profile.

Gene expression in FACS-sorted neutrophils and keratinocytes qRT-PCR analysis in (A–F) neutrophils and in (G–L) keratinocytes from the ears of mice with inducible dual epidermal deletion of c-Jun and junB (DKO*) and triple epidermal deletion of c-Jun, JunB and S100A9 (TKO*) with severe psoriasis-like phenotype (n=4–9). (M) pSTAT3 immunohistochemistry in the ears of DKO* and TKO* mice with severe psoriasis-like phenotype (scale bar=100 µm). (N) Quantification of positive pSTAT3 cells in the epidermis (n=3–4, 4–5 fields per slide).
In contrast, s100a9 mRNA was significantly less induced in keratinocytes isolated from TKO* compared with DKO* (figure 5G), while the increase in s100a8 was comparable (figure 5H). Interestingly, while il-1b was similarly increased in DKO* and TKO* keratinocytes, il-6 and osm mRNA in TKO* were comparable with controls and tnf-a higher (figure 5I–L), suggesting that keratinocyte-derived s100a9 affects epidermal expression of cytokines, such as il-6 and tnf-a, during skin inflammation. STAT3 is an important transcription factor downstream of IL-6 and Oncostatin-M, involved in Ps and other inflammatory diseases.26–28 DKO* and TKO* lesional skin displayed nuclear phosphorylated STAT3 expression, indicating activated JAK/STAT signalling (figure 5M). Importantly, pSTAT3 was higher in TKO* epidermis compared with DKO*, consistent with more severe skin phenotype (figure 5N). Altogether, keratinocyte-derived S100A9 likely modulates epidermal expression of genes potentiating JAK/STAT signalling and skin inflammation, while it reduces local and systemic TNFα production important for joint inflammation.
S100A9 and CP but not S100A8 are potential Ps and PsA markers in humans
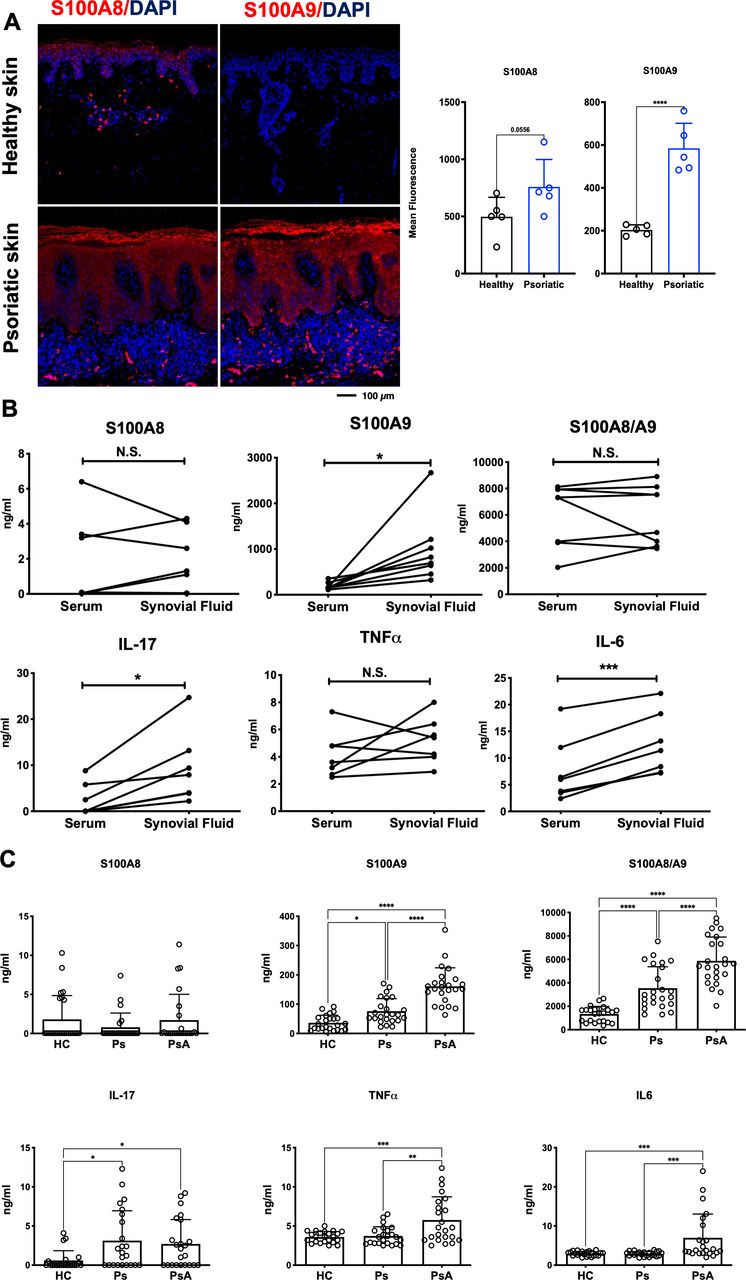
To translate these findings to human disease, we assessed the expression of S100A9 and S100A8 in lesional skin of patients with Ps and measured serum and synovial fluid levels of S100A9, S100A8, CP, IL-17, TNFα, IL-6, VEGF and LCN2 in patients with Ps, patients with PsA and healthy controls (HC). Consistent with increased CP in Ps lesional skin, S100A8 and S100A9 were elevated in hyper-proliferating keratinocytes and infiltrating immune cells of psoriatic plaques, while both proteins were low to undetectable in skin from healthy individuals (figure 6A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Analyses in psoriatic patient samples. (A) S100A8 and S100A9 (red) immunofluorescence staining and quantification in human healthy skin and psoriatic skin (n=5, scale bar=100 µm). (B) S100A8, S100A9, S100A8/A9 (Calprotectin), Interleukin (IL)−17, Tumour necrosis factor alpha (TNF-α) and Interleukin (IL)−6 levels in the serum and synovial fluid of patients with psoriatic arthritis (PsA; n=8, ns=non-significant). (C) S100A8, S100A9, S100A8/A9 (Calprotectin), IL-17, TNF-α and IL-6 serum levels of healthy controls (HC), patients with psoriasis (Ps) or patients with PsA (each group n=24).
Analysis of matched serum and synovial fluid samples from patients with PsA showed overall comparable concentrations between the two biological samples, except for S100A9, IL-17, IL-6 and VEGF that were higher in synovial fluid (figure 6B, online supplemental figure S6A), possibly due to accumulation of immune cells, such as S100A9-expressing neutrophils in the joints. ELISA analyses of sera from a larger cohort of HC, patients with Ps and those with PsA revealed a reasonable correlation between circulating CP or S100A9 homodimers and disease activity scores in patients with Ps and PsA (online supplemental figure S6B). Importantly, S100A9 homodimers and CP were significantly increased in patients with PsA, when compared with Ps and to HC (figure 6C). Furthermore, levels of S100A9 homodimers and CP were increased in patients with Ps compared with HC while no significant differences between groups were observed for S100A8 homodimers (figure 6C). In comparison, while serum IL-17 and LCN2 were higher in patients with Ps and PsA, these could not discriminate between the two groups and high TNFα, IL-6 and VEGF would identify PsA but not Ps (figure 6C, online supplemental figure S6CS6C). These data suggest that S100A9 is a critical mediator in psoriatic skin and joint disease and that S100A9/CP may serve as markers to identify patients with Ps who develop PsA.
Discussion
Epithelial homeostasis is critical for the regulation of inflammation. Here, we show that epithelial expression of the S100A9 alarmin plays a critical role in the severity of psoriatic skin disease and its spreading to the joints. Several studies investigated the role the CP-forming alarmins S100A8 and S100A9 in Ps models. Both proteins are upregulated in the imiquimod (IMQ) model of psoriasiform skin inflammation15 29 and in GEMMs for psoriasis-like disease, such as the DKO*,9 CARD14E138A knock-in,30 K14-Vegfa,31 K14-IL-17A,32 K14-IL-2333 and K5-Stat3C34 bi-transgenics, but functional studies where S100A8 or S100A9 are inactivated in Ps models are still scarce. While one study reported enhanced IMQ-induced skin hyperplasia in S100A8−/− and to a lesser extent in S100A9−/− mice,15 we observed decreased skin thickening in IMQ-treated S100a9−/− mice that was consistent with ameliorated skin and joint disease on global inactivation of S100A9 in the DKO* genetic model.10 While the contradictory results using IMQ could be attributed to different genetic backgrounds and time points of analyses, the complex function of S100 proteins emphasises the need to evaluate cell-specific roles of S100A8, S100A9 and their complexes and to extend the analyses beyond the skin.23
In skin inflammation, S100A9-expressing cells are keratinocytes, neutrophils and macrophages. DKO* mice reconstituted with S100A9−/− bone marrow displayed reduced skin thickening, while transplanting S100A9-proficent bone marrow into DKO*-S100A9−/− mice did not worsen the disease,10 indicating that keratinocytes and immune cells contribute together to Ps-like disease. Evaluating the role of neutrophil-expressed S100A9 in Ps and PsA genetically without resorting to bone marrow chimaeras is difficult since all available GEMMs for Ps and PsA rely on the Cre-lox system. The current study is the first to use a new S100A9 floxed allele to investigate the role of epidermal-derived S100A9 in vivo. One of the most striking observations from the analysis of TKO* mice is that the epidermis contributes very little to circulating S100A9 in psoriatic mice. Hence, there was no difference between DKO* and TKO* when measuring CP or S100A9 homodimers in serum, although S100A9 protein was greatly reduced in TKO* epidermis. This is in stark contrast with DKO*-S100A9−/− mice, where only S100A8 dimers are detected in the serum10 (data not shown). Nevertheless, epidermal-specific inactivation of S100A9 enhanced skin inflammation and increased PsA incidence in TKO*, indicating a regulatory role of epidermal S100A9 in psoriasis-like disease. Expression of S100A9 in keratinocytes seems to modulate the number and activity of skin-infiltrating neutrophils. An increase of 2–3 folds of Ly6B+ myeloid cells and Ly6B-S100A9 double positive cells, most likely neutrophils, was observed in TKO* skin sections, while whole ear proteomics revealed neutrophil activation and degranulation signatures. Interestingly, our previous iTRAQ proteomic comparison of DKO* and DKO*-S100A9−/− epidermis identified immune cell trafficking and activation as one of the most altered pathways.10 Our data thus indicate that neutrophils are likely more abundant and/or more active in the skin of TKO* mice and contribute to more severe local inflammation. How s100a9 gene inactivation in keratinocytes leads to increased immune cell infiltration remains to be clarified, but altered expression of cytokines, such as IL-6 and TNFα, and increased JAK/STAT signalling in S100A9-deficient keratinocytes could provide a first hint.
The DKO* mouse is one of the few psoriasis models displaying features of PsA.35 In contrast to the situation in DKO*-S100A9−/− mice,10 epidermal inactivation of S100A9 led to increased incidence of PsA. PsA severity was, however, comparable between DKO* and TKO*, consistent with comparable levels of circulating S100A9, IL-17A and IL-6. As TNF signalling is essential for joint disease in DKO* mice,9 we postulate that the modest increment in circulating TNFα in TKO* mice, likely originating from increased epidermal tnf-a expression, is one of the factors enhancing PsA incidence. IL-17, IL-6 and S100A9-containing complexes, which are not affected by epidermal inactivation of S100A9, together with S100A9-expressing myeloid cells infiltrating the joints, additionally contribute to bone24 and proteoglycan loss in DKO* and TKO* mice.
In an experimental model for rheumatoid arthritis, S100A9/CP neutralising antibodies had beneficial effects comparable with anti-TNFα,36 the most potent PsA inhibitors in the clinic.37–39 In light of the mouse data, topical therapies aiming at inhibiting S100A9 in the skin might be counterproductive, while systemic inhibition of S100A9/CP with drugs or neutralising antibodies is worth evaluating in GEMMs with Ps and PsA, as a possible complement to anti-TNFα therapies.
Increased CP in serum16 40–42 in skin biopsies10 43 44 and more recently in the stratum corneum 45 has been correlated with disease activity in patients with Ps. We observed that S100A8 dimers were not increased in the serum of patients with Ps and those with PsA, thus S100A8 dimers likely play a minor role. In contrast, circulating CP and S100A9 homodimers were elevated in patients with Ps and even more in patients with PsA. The synovial concentrations of these species were either comparable (CP) or higher (S100A9) than in serum, which would support an active local role of S100A9-containing complexes and S100A9-producing cells in the joints. While serum CP has previously been correlated with PsA severity,8 46 this is the first time that S100A8 and S100A9 dimers are measured along with S100A8/S100A9 CP complexes. We found that S100A9 and CP efficiently discriminated healthy, patients with Ps and patients with PsA. Serum S100A9 and/or CP could therefore help identifying patients with Ps developing PsA. Given that CP is already a validated clinical parameter in inflammatory bowel disease,23 longitudinal assessment in larger patient cohorts with Ps is feasible. Early identification of patients at risk of developing PsA will certainly allow the implementation of better therapies and will advance our understanding of Ps.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study involves human participants and was approved by Institutional Review Board of the University Hospital Erlangen (Ethical approval #157_20 B) Participants gave informed consent to participate in the study before taking part.
Acknowledgments
We are grateful to the CNIO Histopathology, Animal facility, Confocal and Flow Cytometry core units for their support. We thank Fox Chase Cancer Centre Transgenic Facility for the generation of S100A9 floxed mice and J Roth/T Vogel for sharing S100A8 and S100A9 antibodies. We would like to thank members of the former Wagner laboratory at CNIO, Özge Uluckan, Alvaro Ucero, Vanessa Bermeo and Ana Guío-Carrión for advice and technical help and Dr Luis Iñiguez de Onzoño (Hospital General de Albacete, Spain) for pathological evaluation of histological samples.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Thomas Pap
LFM and NG-L contributed equally.
Correction notice This article has been corrected since it published Online First. The author's name, Sergio Oterino-Sogo, has been corrected. Also, the open access licence has been updated to CC BY. 25th May 2023.
Contributors Conceptualisation and methodology: LFM, NGL, LB and EFW. Investigation: LFM, NGL, FS, BB, SR, LB and JTO. Data analysis and/or technical support: DM, SOS and MJ. Writing and editing: LFM, NGL, LB and EFW. Resources: SG, RSP, AR, GS and EFW. Funding acquisition and supervision: EFW. Guarantor: EFW.
Funding The Wagner laboratory at the Medical University of Vienna (MUV) is supported by an ERC‐AdG 2016 CSI‐Fun‐741888, a H2020‐MSCA‐ITN 2019‐859860‐CANCERPREV grant and the MUV. GS and AR are supported by the Deutsche Forschungsgemeinschaft (DFG-FOR2886 PANDORA and the CRC1181 Checkpoints for Resolution of Inflammation). Additional funding was received by the Bundesministerium für Bildung und Forschung (BMBF; project MASCARA), the ERC-SyG 2018 (810316 4D Nanoscope), ERC-STG 2019 (853508 BARRIER BREAK) and the IMI-funded project Hippocrates. The Oxford Laboratory at the Biomolecular Research Centre at Boise State University was supported by the National Institutes of Health, NIGMS P20GM109095 and P20GM103408.
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.