Article Text

Abstract
Objectives Giant cell arteritis (GCA) is the most common primary vasculitis, preferentially affecting the aorta and its large-calibre branches. An imbalance between proinflammatory CD4+ T helper cell subsets and regulatory T cells (Tregs) is thought to be involved in the pathogenesis of GCA and Treg dysfunction has been associated with active disease. Our work aims to explore the aetiology of Treg dysfunction and the way it is affected by remission-inducing immunomodulatory regimens.
Methods A total of 41 GCA patients were classified into active disease (n=14) and disease in remission (n=27). GCA patients’ and healthy blood donors’ (HD) Tregs were sorted and subjected to transcriptome and phenotypic analysis.
Results Transcriptome analysis revealed 27 genes, which were differentially regulated between GCA-derived and HD-derived Tregs. Among those, we identified transcription factors, glycolytic enzymes and IL-2 signalling mediators. We confirmed the downregulation of forkhead box P3 (FOXP3) and interferon regulatory factor 4 (IRF4) at protein level and identified the ineffective induction of glycoprotein A repetitions predominant (GARP) and CD25 as well as the reduced T cell receptor (TCR)-induced calcium influx as correlates of Treg dysfunction in GCA. Inhibition of glycolysis in HD-derived Tregs recapitulated most identified dysfunctions of GCA Tregs, suggesting the central pathogenic role of the downregulation of the glycolytic enzymes. Separate analysis of the subgroup of tocilizumab-treated patients identified the recovery of the TCR-induced calcium influx and the Treg suppressive function to associate with disease remission.
Conclusions Our findings suggest that low glycolysis and calcium signalling account for Treg dysfunction and inflammation in GCA.
- giant cell arteritis
- autoimmunity
- inflammation
- T-lymphocyte subsets
Data availability statement
Data are available upon reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Tregs, displaying reduced suppressive function and increased expression of IL-17, have been implicated in the pathogenesis of giant cell arteritis (GCA).
What does this study add?
Comparative transcriptomic and protein expression analysis of GCA-derived and health blood-donor-derived Tregs identified aberrations of GCA Tregs such as downregulation of transcription factors, glycolytic enzymes as well as low activation-induced calcium signalling and induction of effector molecules.
How might this impact on clinical practice or future developments?
We identify novel pathogenic correlates of GCA activity, which may be useful for monitoring disease activity, especially in tocilizumab-treated patients.
Treg dysfunction may represent a new target for the treatment of GCA.
Introduction
Giant cell arteritis (GCA) is the most common form of systemic vasculitis, affecting the elderly, with a peak incidence at the age of 70–80 years.1 GCA typically involves the aorta and/or its large-calibre branches.2 The localisation and type of affected arteries largely determines the clinical manifestations of GCA, which include cranial symptoms such as headache and masticatory claudication, polymyalgia and non-specific systemic symptoms, that is, fever, night sweat and unintended weight loss. The histological hallmark of GCA is focal granulomatous inflammation.3 Different studies suggested infectious agents, such as herpes simplex virus, varicella zoster virus, parvovirus B19 and Chlamydia pneumoniae as likely disease triggers.4–6 Such infectious agents or an alternative trigger have been suggested to cause abnormal maturation of dendritic cell in the adventitia and the consequent activation of CD4+ T cells.7–9 The Th1-interferon γ (IFNγ) axis and the IL-6-Th17 axis are the main immune responses that dominate the GCA inflammation. While glucocorticoids or tocilizumab (TCZ), a monoclonal antibody against the IL-6 receptor, effectively suppress the IL-6-Th17 axis, the Th1 pathway appears to be less amenable to treatment.8 9
Various autoimmune diseases such as rheumatoid arthritis, multiple sclerosis, systemic lupus erythematosus and systemic sclerosis have been associated with regulatory T cell (Treg) dysfunction.10–12 The forkhead box P3 (FOXP3) is indispensable for the development and function of Treg. Several studies have associated reduced expression of FOXP3 with the loss of immune tolerance and autoimmune inflammation.13–15 Besides FOXP3, the suppressive potential of Tregs critically depends on an array of molecules, which stabilise their polarisation and/or directly mediate their effector functions. Notable examples include the interferon regulatory factor 4 (IRF4), the α chain of the interleukin 2 receptor (IL-2Rα/CD25), the cytotoxic T lymphocyte protein 4 and the glycoprotein A repetitions predominant (GARP).16–22 Genetic variants affecting the function or the expression of these molecules have been reported to underlie monogenic inborn errors of immunity, which cause immune dysregulation18–22 or to confer susceptibility for autoimmune diseases.23–25
An imbalance between proinflammatory CD4+ T helper (Th) cell subsets, that is, Th1 and Th17 cells, and Tregs is thought to be involved in the pathogenesis of GCA. There is scarce evidence regarding the role of regulatory T cells (Tregs) in GCA inflammation. In particular, two studies reported reduced Treg counts in peripheral circulation of patients with GCA, which, however, did not associate with the GCA activity.26 27 A more recent study identified increased Treg counts as a correlate of TCZ-induced remission of GCA.28 Furthermore, Tregs in GCA were reported to display proinflammatory Th17-like properties at the expense of their suppressive function.28 In this study, we aimed to delineate the dysfunction of Tregs in GCA. To this end, we integrated transcriptomic and proteomic data from Tregs, collected from patients with different disease activity and variable immunomodulatory regimens.
Materials and methods
Information on the study population and the experimental methods employed in the present work, including RNA-sequencing, the phenotypic and functional characterisation of regulatory T cells as well as the statistical analysis, is provided in the online supplementary text.
Results
Study population
Studied subjects characteristics are summarised in table 1. Information on GCA patients’ disease activity status and treatments at blood sampling is provided in online supplemental table 1.
Supplemental material
Characteristics of studied subjects at blood sampling
Transcriptomic profiling of GCA Tregs
First, we performed differential transcriptome analysis between Tregs from patients with GCA (n=12; active disease, n=6, in remission, n=6; see online supplemental table 2 for patients’ characteristics) and healthy blood donors (HD, n=6). Pairwise comparison of active GCA versus HD-derived Tregs, using adjusted p value <0.05 and cut-off fold change >1.47 (log2FC=0.56), identified 27 differentially expressed genes (DEGs) (figure 1A). Among DEGs, we highlighted an enrichment for genes related to three molecular classes: Treg transcription factors (FOXP3, IRF4 and IKZF4), glycolytic enzymes (ENO1, PFKP, LDHA) and molecules downstream to IL-2 signalling (CISH, SOCS2). Furthermore, relative quantification showed an overall lower expression of these transcripts in GCA Tregs, especially in the active cases, as compared with healthy Tregs (figure 1B). To evaluate the influence of glucocorticoids on the observed differences in transcript expression, we reanalysed transcriptome data after classifying patients with GCA (both active and inactive) into glucocorticoid-receiving (n=7) and those without glucocorticoid treatment (n=5). This identified no significant differences (CISH: p value=0.5025; ENO1: p value=0.3308; FOXP3: p value=0.9773; IKZF4: p value=0.7096; IRF4: p value=0.7096; LDHA: p value=0.6010; PFKP: p value=0.7424; SOCS2: p value=0.7096), suggesting that differential transcript expression by GCA Tregs was independent of the treatment with glucocorticoids.

Transcriptional, metabolic, and signalling disturbances in GCA Tregs. (A) Volcano plot showing differentially expressed genes between active GCA Tregs vs healthy Tregs (adjusted p value<0.05, log2(fold change)>0.56). (B) Heatmap analysis showing differential expression of selected genes encoding transcription factors, glycolytic molecules, and IL-2/STAT-5 signalling pathway, in Tregs from different groups (active GCA cases, GCA in remission, healthy donors). Treatment of each studied patient is indicated: CS, corticosteroids; csDMARDs, conventional synthetic disease-modifying antirheumatic drug; GCA, giant cell arteritis; TCZ, tocilizumab.
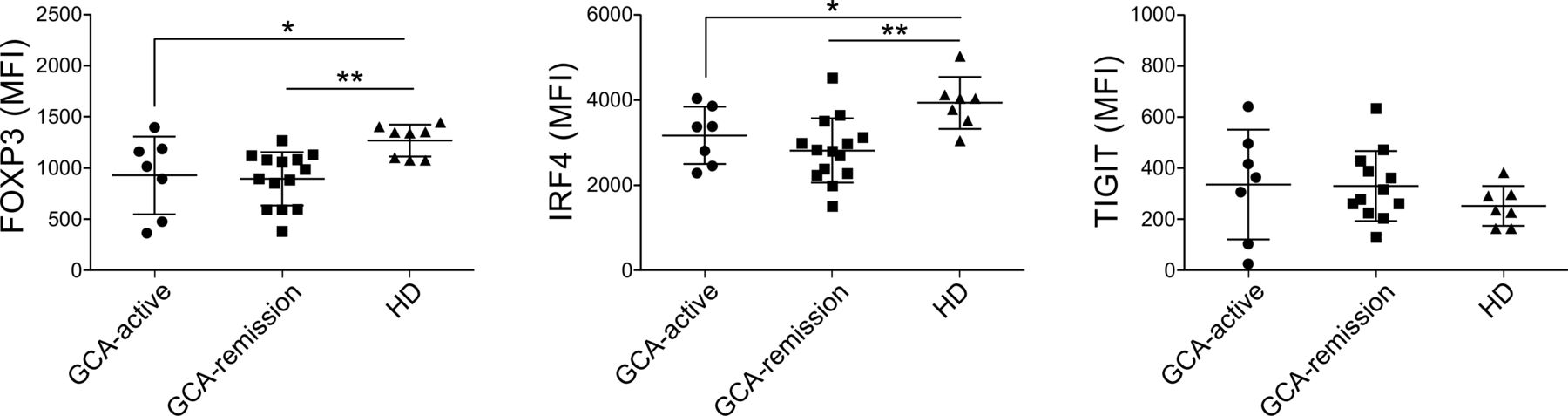
Differential expression of FOXP3 and IRF4 at the level of transcript was evaluated at protein level by flow cytometry (figure 2). In line with the transcriptome data, both FOXP3 and IRF4 levels were lower in GCA Tregs than HD Tregs. Lower expression levels of FOXP3 in GCA Tregs did not associate with significant differences in FOXP3-positivity within CD4+ CD25hiCD127lo Tregs (online supplemental figure 1). Treg from patients in remission and those with active GCA displayed similar expression levels of FOXP3 and IRF4. On the other hand, TIGIT, whose transcript levels were reduced in most GCA samples, displayed similar expression among different groups of patients and HD.

Reduced expression of FOXP3 and IRF4 by GCA Tregs, measured by flow cytometry as MFI. Bars represent the means±SDs. GCA, giant cell arteritis; HD, healthy blood donor; MFI, median fluorescence intensity.
Treg dysfunction in GCA
Despite the fact that patients with GCA and HD displayed similar CD4+ CD25hiCD127lo Treg counts (figure 3A), we identified several qualitative abnormalities with respect to the expression of effector molecules by GCA Tregs. GARP is involved in TGF-β maturation and the suppressive potential of Tregs both in vitro and in vivo depends on its expression.19 29 After 18 hours of CD3/CD28 stimulation with Dynabeads, GCA Tregs expressed significantly lower levels of GARP than HD Tregs (figure 3B). The suppressive function of Tregs critically depends on CD25, whose reduced expression by Tregs has been linked to diverse autoimmune diseases.30 31 Similar to GARP, GCA Tregs displayed impaired induction of CD25 after CD3/CD28 stimulation (figure 3C). However, we observed no difference in GARP or CD25 expression between Tregs from active and inactive GCA. TCR-induced calcium signalling has been linked to both Treg development and suppressive function.32 Here, we identified a marked reduction in immediate TCR-induced calcium influx in GCA Tregs, especially in those from patients with active disease (figure 3D,E). In contrast, the difference in calcium flux between GCA Tregs in remission and healthy Tregs was not statistically significant (p value=0.1936). It has been demonstrated that the exon 2 of FOXP3 physically binds RORγT to prevent Th17 polarisation, and that patients with GCA display a higher frequency of IL-17 producing and FOXP3-exon 2 deficient (FOXP3∆2) Tregs, which could play a pathogenic role in GCA.28 We were able to recapitulate these observations in our cohort (figure 3G–I). In addition, we observed that at single cell level, FOXP3Δ2 Tregs expressed less CD25, as compared with their FOXP3 exon 2-expressing counterparts. To evaluate the relevance of identified phenotypic abnormalities of GCA Tregs, we performed a suppression assay evaluating the proliferation of conventional T cells (CD4+ CD25loCD127hi) in the presence or absence of CD4+ CD25hiCD127lo Tregs (figure 3H). The results show significantly reduced suppressive potential of GCA Tregs. Furthermore, the fact that the suppressive potential of Tregs from active patients was significantly lower to the one of patients in remission, suggests the association of Treg dysfunction with disease activity. Differences in differentiation state of Tregs could account for Treg dysfunction in GCA. To evaluate this, we measured the frequencies of activated Tregs and resting Tregs, as those were defined by Miyara et al.33 In line with previous reports,27 28 the latter revealed comparable proportions of activated to resting Tregs between GCA patients and HD (online supplemental figure 2), suggesting the occurrence of Treg dysfunction in the presence of normal Treg population dynamics.

Treg dysfunction in GCA. (A) Frequencies of Tregs (CD4+CD25hiCD127lo) in different groups (active GCA, GCA in remission, healthy donors). (B) Protein expression of GARP after 18 hours of CD3/CD28 stimulation. (C) Protein expression of CD25 at rest and after 18 hours of CD3/CD28 stimulation. (D) Representative calcium flux tracing (GCA patients vs healthy donors). At 60 s, anti-CD3 (clone: OKT3) was added (5 µg/mL). At 180 s, CaCl2 was added (7 mM). At 330 s, ionomycin was added (14 µg/mL). (E) Calcium flux in Tregs (normalised to baseline) after CaCl2 addition. (F) Representative gating strategy to quantify FOXP3Δ2 cells, using anti-FOXP3 clone 236A/E (total FOXP3) and clone 150D (measuring only exon 2). (G) Frequencies of FOXP3Δ2 cells (% Tregs). (H) Single-cell expression level of CD25 in FOXP3Δ2 Tregs versus FOXP3 exon 2-expressing Tregs at the basal state. (I) Frequencies of IL-17 producing Tregs (IL-17A+) (% Tregs, CD4 +CD25 hi CD127lo). (J) Treg suppression assay, conventional T cell proliferation in the presence of autologous Tregs: representative plots from patients with GCA (active and in remission) and a HD are shown. (K) Summary of cell proliferation normalised to the positive control of each sample (without Tregs) in patients with GCA (GCA-active: n=5, GCA-remission: n=10) and HD (n=5). GCA, giant cell arteritis; HD, healthy blood donor; MFI, median fluorescence intensity.
Glycolysis inhibition recapitulates dysfunction of GCA Tregs
As presented above, transcriptome analysis revealed lower expression of glycolytic enzymes, such as phosphofructokinase (PFKP) and enolase 1 (ENO1), in GCA Tregs (figure 1B). Despite several controversies regarding Treg metabolism, recent studies have shown that glycolysis promotes FOXP3 expression, and under certain circumstances, the suppressive function of human Tregs.34 35 Therefore, to evaluate the likely role of reduced glycolysis in human Treg dysfunction, we evaluated the effect of glycolysis inhibition on Treg phenotypes, using 2-deoxyglucose (2-DG). Glycolysis inhibition in healthy Tregs led to failure of GARP and CD25 upregulation after 18 hours of TCR stimulation (figure 4A,B). Furthermore, TCR-induced calcium influx was effectively abolished by glycolysis inhibition (figure 4C,D). These findings suggests a direct link between glycolysis and calcium signalling in human ex vivo Tregs. On the other hand, glycolysis inhibition in GCA Tregs had no significant additive effect on reduced upregulation of GARP or CD25 (online supplemental figure 3A,B). Similar was the case with TCR-induced calcium influx in Tregs from patients with active GCA (online supplemental figure 3C). Finally, as glycolysis has also been linked to alternative splicing of FOXP3 in human iTregs,36 we tested whether glycolysis inhibition in healthy Tregs could lead to higher frequencies of FOXP3Δ2 Tregs, which was the case (figure 4E). Similar to ex vivo GCA Tregs, FOXP3Δ2 cells expressed less CD25 than FOXP3 exon 2-expressing Tregs (figure 4F).

Glycolysis inhibition of healthy Tregs. (A) Protein expression of GARP after 18 hours of CD3/CD28 stimulation, following 48 hours of pre-incubation with 2-deoxyglucose (2-DG) (2 mM). (B) Protein expression of CD25 at rest and after 18 hours of CD3/CD28 stimulation, following 48 hours of 2-DG (2 mM). (C) Representative calcium flux tracing. Sorted Tregs were analysed in the presence or absence of 2-DG (50 mM). Anti-CD3, CaCl2, and ionomycin were added at timepoints as described for figure 3D. (D) Calcium flux in Tregs (normalised to baseline) after CaCl2 addition. Two concentrations of 2-DG were used for glycolysis inhibition (50 mM and 2 mM). (E) Frequencies of FOXP3Δ2 cells (% Tregs). (F) Single-cell expression level of CD25 in FOXP3Δ2 Tregs versus FOXP3 exon 2-expressing Tregs at the basal state, with and without 2-DG (2 mM). GARP, glycoprotein A repetitions predominan; MFI, median fluorescence intensity.
TCZ partially normalises GCA Treg dysfunction
As TCZ has been shown to enhance the suppressive function of Treg in GCA,28 we evaluated the previously identified phenotypical changes in GCA Tregs in the subgroup of TCZ-treated patients, which all were in remission. As shown in figure 5A,B, TCZ treatment appears to enhance IRF4 but not FOXP3 expression in Tregs. Furthermore, the induction of GARP and CD25 remained impaired, also in Tregs from TCZ-treated patients (online supplemental figure 4). Similar to the rest of patients in remission, treatment with TCZ appears to normalise TCR-induced calcium influx in Tregs (figure 5C). Finally, we were able to recapitulate the previously described reduction in the frequency of FOXP3Δ2 Tregs in TCZ-treated patient with CGA (figure 5D).28 Partial reversion of Treg dysfunction by TCZ, is suggested by the improved suppressive potential of Tregs in the context of the suppression assay (figure 5E). Altogether, these findings demonstrate that IL-6 receptor blockade seems to improve calcium signalling and the suppressive function of Tregs in GCA. Most evaluated Treg parameters, especially TCR-induced calcium influx and the suppressive function, whose recovery correlates with remission, were comparable in GCA remission with or without TCZ (figure 5B–E). This together with the similar results after performing a subanalysis of patients in remission with or without methotrexate (online supplemental figure 5), suggest that improved function of Tregs is rather the consequence of effective immunomodulation in GCA and not a medication-specific effect.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Tocilizumab normalised IRF4, FOXP3Δ2, and calcium flux. (A) Protein level of IRF4 in Tregs. (B) Protein level of FOXP3 in Tregs. (C) Frequencies of FOXP3Δ2 (% Tregs). (D) Calcium flux in Tregs (normalised to baseline) after CaCl2 addition. (E) Treg suppression assay, conventional T cell proliferation in the presence of autologous Tregs: summary of cell proliferation normalised to the positive control of each sample (without Tregs) in patients with GCA (GCA-active: n=5, GCA-remission-TCZ: n=5, GCA-remission-TCZ: n=5) and HD (n=5). GCA, giant cell arteritis; HD, healthy blood donor; TCZ, tocilizumab.
Discussion
Here we explored the aetiology of Treg dysfunction in GCA and identified for the first time the downregulation of IRF4 and FOXP3, which are critical transcription factors for both the polarisation and suppressive functions of Tregs, the reduction of TCR-induced calcium signalling as well as the insufficient upregulation of effector molecules as causes of Treg pathogenicity in GCA. Further, using transcriptome analysis we identify a marked downregulation of glycolytic enzymes in GCA Tregs, which may play a central role in the aetiology of most identified Treg dysfunctions.
Imbalances in the expression of FOXP3 isoforms have been reported in various autoimmune diseases, including rheumatoid arthritis and autoimmune thyroiditis.37 Among FOXP3 isoforms, expression of the exon 2 containing FOXP3 appears critical for the suppressive function of human iTregs.36 The fact that patients with FOXP3 variants selectively affecting the expression of exon 2 develop IPEX, provides additional evidence on its in vivo regulatory function.38 Mechanistically, based on murine Treg findings, it has been suggested that the regulatory role of the exon 2 of FOXP3 can be explained by the fact that it physically antagonises RORγT, and exon 2-skipping increases the propensity of Tregs to produce IL-17, which potentially exacerbates inflammation.39 40 The finding by Miyabe et al that GCA patients display higher frequencies of FOXP3∆2 Tregs, which we confirmed, proved evidence on the pathogenicity of alternative FOXP3 splicing and especially reduced expression of the exon 2 of FOXP3 in GCA.28
TCR-induced calcium signalling largely depends on the pathway of store-operated calcium entry (SOCE).41 Conditional deletion of SOCE mediators in murine Tregs and the consequent loss of TCR-induced calcium influx, affected both their polarisation and effector differentiation, resulting in systemic autoimmunity.32 Human STIM1 deficiency abrogates TCR-induced calcium influx and besides immunodeficiency causes autoimmunity,42 which given the role of SOCE in murine Tregs could be explained by Treg dysfunction. Here we identify reduced TCR-induced calcium influx in GCA Tregs, which appeared to normalise in Tregs from TCZ-treated and the rest of patients in remission, suggesting the direct correlation of this finding with GCA inflammation.
Despite the longstanding belief that Tregs are not glycolytic but rather rely on the oxidative pathway of glucose metabolism,43 35 several recent studies have identified various aspects of human Treg biology, including FOXP3 alternative splicing, cell migration, proliferation and IL-2 signalling, which all depend on glycolysis.35 36 43–46Likewise, reduced glycolysis and the consequently compromised suppressive potential of Tregs has been implicated in pathogenesis of autoimmune diseases, such as multiple sclerosis and type 1 diabetes mellitus.36 47 Mechanistically, de Rosa et al have shown that glycolysis controls FOXP3 splicing and enhances the expression of exon 2-containing FOXP3, which is involved in the suppressive activity of Tregs.36 Furthermore, a murine T cell study reported that the glycolytic metabolite phosphoenolpyruvate enhances TCR-induced calcium influx and that calcium mobilisation in T cells was reduced after 2-DG-mediated inhibition of glycolysis.48 In this study, side-by-side characterisation of GCA Tregs and in vitro glycolysis-inhibited Tregs demonstrate that GCA-associated Treg abnormalities, such as the increased frequency of FOXP3∆2 Tregs and the reduced TCR-induced calcium influx, can be recapitulated by glycolysis inhibition in healthy Tregs. Considering the central role of exon 2-expressing FOXP3 and the direct link between glycolysis and calcium signalling in Tregs, which we identify in the present study, our findings suggest that the downregulation of glycolytic enzymes in GCA Tregs is a central event in the aetiology of Treg dysfunction in GCA.
Longer term follow-up of GiACTA trial revealed that a minority of treated patients, that is, 42%, maintained clinical remission after stopping treatment with TCZ.49 The requirement of long-term treatment together with the limited reliability of acute phase reactants under TCZ treatment can render the monitoring of disease activity expensive, necessitating vascular imaging such as PET.50 Therefore, laboratory biomarkers reflecting disease activity independently of the acute phase response may be useful for evaluating disease activity in TCZ-treated patients. Our findings suggest the TCR-calcium response and the expression of IRF4 by Tregs as markers of GCA remission in TCZ-treated patients.
Apart from the above-mentioned correlates of remission, GCA Tregs – even from TCZ-treated patients – still display a largely dysfunctional phenotype, including lower activation-induced expression of CD25 and GARP. This suggests the need for novel therapeutic approaches with a broader effect on Treg dysfunction. On the other hand, the fact that TCZ or csDMARD-induced remission associated with normalised calcium influx only, highlights the central pathogenic role of compromised calcium signalling in GCA Tregs. The lack of steroid-sparing effect of the calcineurin inhibitor cyclosporine in GCA51 may stem from the critical role of calcium signalling for Treg function and comes in line with the aforementioned finding.
The strengths of our study include the analysis of CD4+ CD25hiCD127lo cells as this gating strategy reliably distinguishes ex vivo Tregs,52 53 and the phenotypic analysis of ex vivo unprimed Tregs, which better reflects the in vivo setting. The use of RNA-Seq for transcriptomic profiling has been suitable to enumerate pathologies in GCA Tregs, which we validated at protein level and experimentally with in vitro analysis of healthy Tregs. Our patient cohort was representative of GCA, including patients in remission and active cases as well as treatment naïve patients. Weaknesses of the study include the small number of tested patients, especially for the transcriptome analysis, where a bigger number of patients could potentially have led to the detection of more DEGs. Study of a larger number of patients with GCA, including pretreatment samples as well as samples from patients with same disease activity status, receiving similar treatment, may have aided evaluation of the possible differential effects of the immunomodulatory agents on the Treg phenotype. On the other hand, small sample size can have underpowered the detection of differences between TCZ-treated patients and HD and may also account for the identification of normal Treg frequency in GCA, in the present study, which deviates from the reported reduced Treg counts in some of the previous studies on GCA Tregs.26 27 In addition, we have not demonstrated the metabolic disturbance directly with a metabolic assay, most well-established of which is the Seahorse metabolic flux assay, due to limitations both regarding the assay sensitivity and the cell availability.54 Another point that requires further research is the characterisation of Tregs from inflamed arteries, whose study would necessitate fresh samples and/or the development staining protocols reliably identifying Tregs and the expression levels of key tolerogenic molecules.
In summary, we present novel abnormalities of Treg function in GCA, suggesting the pathogenic role of low glycolysis and calcium signalling in GCA Tregs. Our findings may aid the development of therapeutic approaches targeting Treg dysfunction in GCA and provide new correlates of disease remission, which may be useful for monitoring disease activity, especially in case of TCZ-treated patients.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Ethics approval
This study was conducted in accordance with the Declaration of Helsinki and was also approved from the Ethical committee of the Hannover Medical School (approval number: 8875). All patients signed an informed consent form.
Acknowledgments
We thank MHH Core Facility Cell Sorting (Dr. Matthias Ballmaier) for Tregs FACS sorting, Core Facility Genomics (Ms Heike Schneider and Mr Torsten Glomb) for RNA-Seq (library preparation, sequencing run, bioinformatics) and Dr Fatih Noyan and Mrs Sabine Buyny for advice with respect to establishing the Treg suppression assay. We sincerely thank giant cell arteritis patients and volunteers for participating in the study, and clinic nurses for their excellent help with blood sampling.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
Contributors Conception and design of the works: GS, IRA. Data acquisition, analysis, interpretation: IRA, GS, OD-B, PG, MK. Patient recruitment: SH, LMR, PG, TW, GS. First manuscript draft: IRA, GS. Funding: GS, FA, IRA, TW, RES. All authors revised and approved the manuscript.
Funding This project was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy—EXC 2155 'RESIST'—Project ID 39087428, the German Federal Ministry of Education and Research (BMBF) through a grant to the German Auto-Immunity Network (GAIN), grant code 01GM1910E Hannover and the Rosemarie-Germscheid Foundation. IRA was supported by the German Academic Exchange Service (DAAD, personal reference number 91720367) and the Hannover Biomedical Research School (HBRS)—Center for Infection Biology (ZIB).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.