Article Text

Abstract
Objectives To determine if the polymorphism encoding the Arg206Cys substitution in DNASE1L3 explains the association of the DNASE1L3/PXK gene locus with systemic lupus erythematosus (SLE) and to examine the effect of the Arg206Cys sequence change on DNASE1L3 protein function.
Methods Conditional analysis for rs35677470 was performed on cases and controls with European ancestry from the SLE Immunochip study, and genotype and haplotype frequencies were compared. DNASE1L3 protein levels were measured in cells and supernatants of HEK293 cells and monocyte-derived dendritic cells expressing recombinant and endogenous 206Arg and 206Cys protein variants.
Results Conditional analysis on rs35677470 eliminated the SLE risk association signal for lead single-nucleotide polymorphisms (SNPs) rs180977001 and rs73081554, which are found to tag the same risk haplotype as rs35677470. The modest effect sizes of the SLE risk genotypes (heterozygous risk OR=1.14 and homozygous risk allele OR=1.68) suggest some DNASE1L3 endonuclease enzyme function is retained. An SLE protective signal in PXK (lead SNP rs11130643) remained following conditioning on rs35677470. The DNASE1L3 206Cys risk variant maintained enzymatic activity, but secretion of the artificial and endogenous DNASE1L3 206Cys protein was substantially reduced.
Conclusions SLE risk association in the DNASE1L3 locus is dependent on the missense SNP rs35677470, which confers a reduction in DNASE1L3 protein secretion but does not eliminate its DNase enzyme function.
- autoimmunity
- lupus erythematosus
- systemic
- polymorphism
- genetic
- scleroderma
- systemic
- arthritis
- rheumatoid
Data availability statement
Data are available upon reasonable request. The dataset analysed was from a publication by two of our authors (CDL, HCA, DSCG et al. Transancestral mapping and genetic load in systemic lupus erythematosus. Nat Commun 2017;8:16021. doi: 10.1038/ncomms16021 (published Online First: 2017/07/18)), and requests for SLE Immunochip data should be sent to CLangefe@wakehealth.edu.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
The coding polymorphism rs35677470 is the causal variant for rheumatoid arthritis (RA) and scleroderma/systemic sclerosis (SSc) association in the DNASE1L3/PXK locus and is reported to eliminate DNASE1L3 enzyme function.
What does this study add?
The coding polymorphism rs35677470 is the causal variant for the systemic lupus erythematosus (SLE) risk association in the DNASE1L3/PXK locus.
Secretion of DNASE1L3 protein is drastically reduced by the 206Cys protein variant.
The DNASE1L3 206Cys variant retains DNase enzyme activity.
How might this impact on clinical practice or future developments?
Patients with SLE, RA and SSc carrying the rs35677470 risk allele are candidates for DNASE1L3 replacement therapy or potential therapies rescuing cellular secretion of the DNASE1L3 protein.
Introduction
DNASE1L3 possesses endonuclease enzyme activity for lipid-associated DNA and nucleosomal chromatin molecules,1 2 which enables it to prevent harmful anti-DNA autoimmune responses towards self-DNA.2 3 Consequently, there is a high penetrance of disease in humans homozygous for a complete loss-of-function mutation in the DNASE1L3 gene.4–7 Common variation in the DNASE1L3 locus is also associated with risk for systemic lupus erythematosus (SLE), scleroderma/systemic sclerosis (SSc) and rheumatoid arthritis (RA). The rs35677470 single-nucleotide polymorphism (SNP) encodes an arginine to cysteine substitution at position 206 of the DNASE1L3 protein (Arg206Cys). This missense variant is known as the causal variant for association of RA and SSc risk with the DNASE1L3 locus in genome-wide (and Immunochip-wide) association studies (GWAS).8–11 The 3p14.3 locus containing the DNASE1L3 gene is associated with risk for SLE in GWAS; however, this association is generally known by the location of a lead associated SNP in a neighbouring gene, PXK.12–14 Functionally, the Arg206Cys variant is reported to eliminate the enzyme activity of DNASE1L32 15 via destabilisation of the protein architecture.16
Materials and methods
Supplemental material
Results
Conditional analysis on rs35677470 eliminates the SLE risk signal in the DNASE1L3/PXK locus
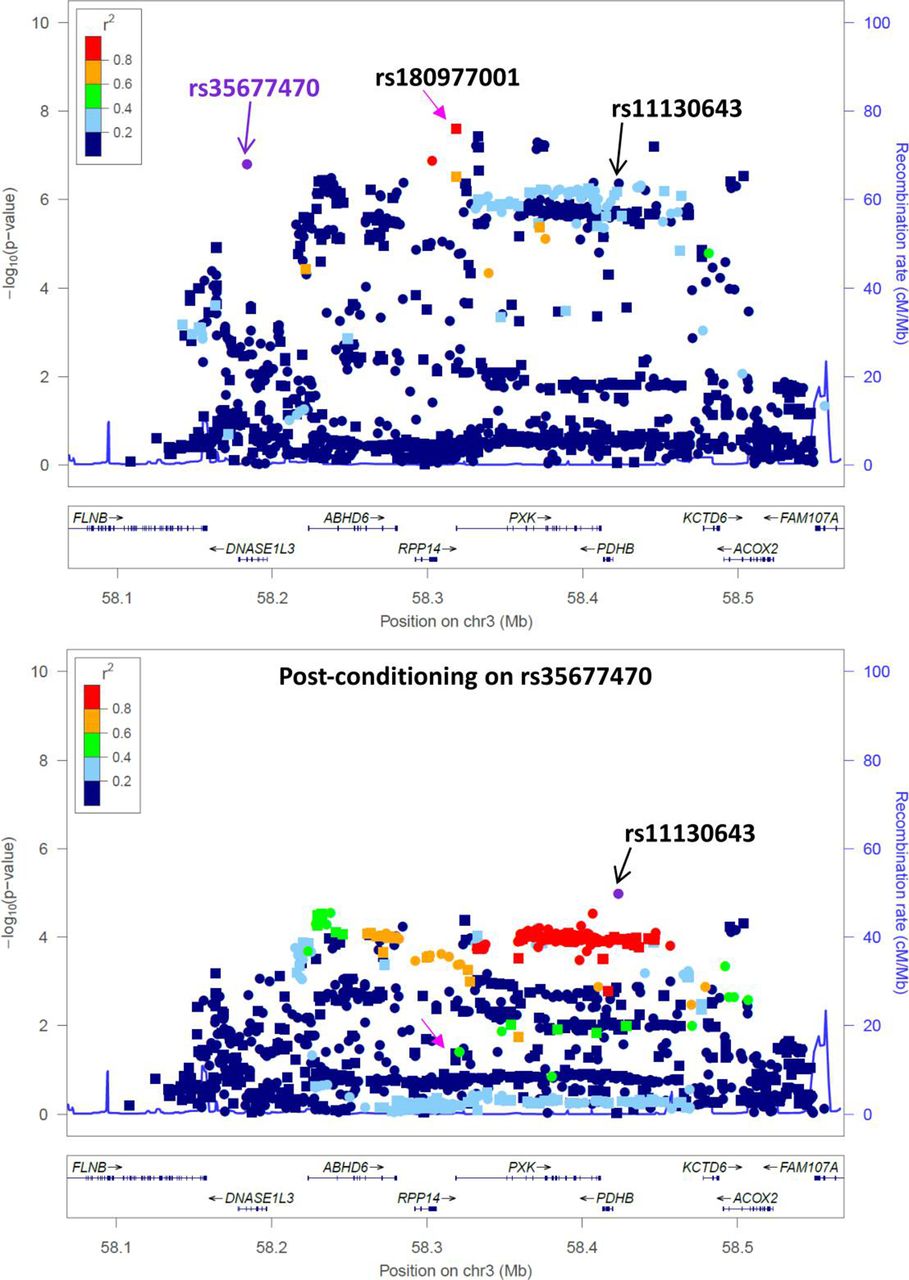
To determine the dependence of the lead risk association signal on the DNASE1L3 Arg206Cys variant, we performed conditional analysis for rs35677470 in the Immunochip association dataset of SLE.13 No significant risk signal remains for the lead SNPs rs180977001 (imputed SNP, P=2.5×10−8 OR=1.27 (1.17–1.39), Pcond =0.043) and rs73081554 (directly genotyped SNP, P=1.3×10−7 OR=1.26 (1.15–1.37), Pcond =0.144) in the region, following adjustment for rs35677470 (figure 1). A residual protective association signal remains, however, following conditioning on rs35677470 (rs11130643 P=4.2×10−7 OR=0.89 (0.85–0.93), Pcond =1×10−5 OR=0.81 (0.74–0.89)(see online supplemental tables S1 and S2).17

The missense variant rs35677470 encoding DNASE1L3 Arg206Cys explains the risk association signal at 3p14.3. A regional association plot for SLE cases with European ancestry from the Immunochip association study13 shows an extended region of association spanning from DNASE1L3 to ACOX2 on chromosome 3. The lead imputed SNP rs180977001 (indicated by a pink arrow in both plots) did not retain any significant signal when analyses were conditioned on rs35677470 (purple symbol in top plot). A residual signal for a protective secondary association remained following rs35677470 conditioning, lead SNP rs11130643 (indicated by a black arrow in both plots). Directly genotyped SNPs are depicted by circles and imputed SNPs are depicted by squares.
We calculated that the SLE OR for heterozygous carriers of the rs35677470 risk allele is 1.14 (1.05–1.24) and the SLE OR for a homozygous risk allele carrier is 1.68 (1.14–2.47), implying that the defect conferred by Arg206Cys is much less deleterious than complete DNASE1L3 deficiency (online supplemental table S3). Hence, we investigated the defect caused by the substitution of cysteine for arginine in DNASE1L3 at position 206.
206Cys variant impedes secretion of DNASE1L3 protein transiently expressed in HEK293 cells, but enzymatic activity is retained
In assaying the cell-free supernatants of HEK293 cells transfected with DNASE1L3 expression constructs, we observed that wild-type rDNASE1L3 206Arg protein was secreted into supernatants, but the protein was not secreted in the presence of the 206Cys encoded by the adenine risk allele of rs35677470 (figure 2A). The difference in secretion appeared despite similar cellular expression (figure 2A,B) of the protein and equal expression of the transcripts (data not shown). Secretion of rDNASE1L3 protein could be blocked with brefeldin A, which inhibits Golgi-mediated protein secretion, indicating that secretion occurs via the classical endoplasmic reticulum Golgi complex secretion pathway as reported (figure 2B,C).1 Blocking of rDNASE1L3 protein secretion led to significant accumulation of intracellular rDNASE1L3. Not surprisingly, the large difference in protein secretion correlated with a large difference in DNASE1L3 endonuclease activity for exogenous plasmid DNA substrate, as well as nuclear chromatin DNA, by the supernatants (figure 2D–H). Notably, a faint band of secreted rDNASE1L3 206Cys protein was sometimes present in supernatants (figure 2B,E). This rDNASE1L3 206Cys supernatant was enzymatically active, resulting in a twofold to fivefold reduction of plasmid DNA and a twofold to fourfold increase in fragmentation of endogenous and exogenous nuclear chromatin (figure 2D–H).

DNASE1L3 protein secretion is abolished by the autoimmune disease risk allele encoding DNASE1L3 206Cys. (A) rDNASE1L3 206Arg protein is detected in the supernatants of HEK293 cells transiently expressing the DNASE1L3 gene from 1-day post-transfection, but rDNASE1L3 206Cys protein is not. Independent western blots were probed with monoclonal and polyclonal antibodies to DNASE1L3. Note that the polyclonal DNASE1L3 antibody detects a non-specific band from FCS at ~25 kDa (not shown). (B) Secretion of rDNASE1L3 from HEK293 cells is inhibited by the Golgi blocker BFA. HEK293 cells were transfected with expression vectors of DNASE1L3 206Arg and 206Cys in the absence of FCS. Thirty hours after transfection, secretion was blocked by the addition of BFA. Cells and cell-free supernatants were harvested 16 hours after treatment with Golgi block and assayed by western blot. (C) rDNASE1L3 protein concentrations quantified by ELISA in HEK293 cell-free supernatants and cell lysates (corrected for cell number) correlates with rDNASE1L3 protein detected by western blot in B. rDNASE1L3 concentrations in cell lysates were measured in a single ELISA well, and concentrations in supernatants are represented as the mean and SD of duplicate ELISA wells. (D) Endonuclease activity of recombinant DNASE1L3 in supernatants correlates with the amount of protein secreted. Enzyme activity of secreted rDNASE1L3 in supernatants (as in B,C) was measured in an endonuclease activity assay using plasmid DNA, or plasmid DNA coated with liposomes, as a substrate. Plasmid DNA was measured by a quantitative PCR (qPCR) assay for GFP. DNase I was spiked into cell-free supernatants of untransfected cells where indicated. (E) HEK293 cells were transfected with 500, 50 or 5 ng of expression vectors of DNASE1L3 206Arg and 206Cys as in B, and rDNASE1L3 protein secretion was assayed by western blot using a polyclonal antibody. (F) rDNASE1L3 DNA enzyme activity was tested in a nuclear chromatin fragmentation assay. Cell supernatants were incubated with isolated nuclei and genomic DNA was isolated. DNA fragmentation was observed by gel electrophoresis. Conditioned supernatants in digestion assay were as follows: lane 1, control vector; lanes 2–4, DNASE1L3 206Arg vector at 500, 16 and 0.5 ng transfections, respectively; lanes 5 and 6, DNASE1L3 206Cys vector at 500 and 16 ng transfections. (G) Quantitative analysis of rDNASE1L3 206Arg and 206Cys nuclear chromatin fragmentation activity. Supernatants from transfected cells in E were combined with purified nuclei. Genomic DNA was isolated and DNA fragmentation was assayed by qPCR as in F. Bars and error bars represent mean values and SD of qPCR assays run in triplicate. Each figure is representative of two (C), three (F–H) or four independent experiments. (H) Fragmentation of cell-free DNA in supernatants of cells transfected in E was measured by a qPCR assay differentiating the ratio of polynucleosomal fragment lengths (360 bp and greater) to fragments of mononucleosomal length (180 bp) and greater. BFA, brefeldin A; cfDNA, cell-free DNA; Ctrl, control; FCS, fetal calf serum; GFP, green fluorescent protein gene; rDNASE1L3, recombinant DNASE1L3 protein; SN, supernatant.
206Cys variant impedes secretion of endogenous DNASE1L3 protein in healthy subjects homozygous for the rs35677470 risk allele
It is reported that high levels of DNASE1L3 transcript are expressed in dendritic cells (DCs) derived from monocytes following culture with GM-CSF and interleukin-4,18 19 and that these cells secrete detectable levels of DNASE1L3 protein.19 We obtained fresh blood from consented healthy volunteers and confirmed the highest expression of DNASE1L3 transcript is present in monocyte-derived dendritic cells (moDCs) (figure 3A). We also confirmed the secretion of DNASE1L3 protein from moDCs is dependent on the classical Golgi complex secretion pathway (figure 3B,C). DNASE1L3 gene expression in moDCs did not differ based on rs35677470 genotype (figure 3A). Furthermore, no difference in cytotoxicity, morphology, quantity or viability of moDCs was observed between rs35677470 genotypes (online supplemental figure S1, data not shown).

{kind=link}
{kind=link}
{kind=link}
Significantly reduced secretion of endogenous DNASE1L3 206Cys from monocyte-derived DCs of homozygous risk allele carriers. (A) Gene expression of DNASE1L3 is greatest in monocyte-derived DC, in comparison with monocyte-derived Mφ, and PBMCs, DCs and CD14+ monocytes. DNASE1L3 expression measured by qPCR (normalised to HPRT1); each symbol is representative of one healthy subject. The legend indicates the genotypes of each subject; black/grey circles are subjects not genotyped. MoDCs secrete DNASE1L3 protein, which accumulates intracellularly when secretion is blocked by BFA. MoDCs were suspended in serum-free RPMI and further cultured for 24 hours (±BFA treatment overnight). DNASE1L3 protein was measured by ELISA in supernatants (B) and cell lysates (C) of healthy subjects homozygous for 206Arg (n=13), homozygous for 206Cys (n=3) or heterozygous (n=3 or 5). (D) Cell-free DNA fragmentation in these moDC supernatants was estimated by a nucleosomal fragmentation qPCR assay as in 2F (n=10 GG subjects in +BFA group). Fragmentation of cell-free DNA in plasma and by plasma was measured by the nucleosomal chromatin fragmentation qPCR as in 2F-H. A further statistical comparison of all 3 genotype groups without Golgi block was performed by ordinary one-way analysis of variance (P=0.0499). Cell-free DNA fragmentation in plasma assayed directly (E) was from healthy subjects homozygous for 206Arg (n=19), homozygous for 206Cys (n=3) or heterozygous (n=9). Plasma combined with purified nuclei in a nuclear chromatin digest assay was from healthy subjects homozygous for 206Arg (n=19), homozygous for 206Cys (n=3) or heterozygous (n=9). Genomic DNA was isolated from digested nuclei and analysed by qPCR (F) and gel electrophoresis (G,H). Each unique unrelated subject is indicated by a symbol, and the mean of each group is represented by the bar. Comparisons for statistical significance were performed where indicated between groups without Golgi block. Rs35677470 genotype groups ‘GG’ or ‘AG’ were compared with the ‘AA’ group using a one-tailed Mann-Whitney test. Statistical significance is given or * denotes a p value of <0.05 or *** denotes a p value<0.001. Each symbol in A–F represents the average value of sample replicates run in triplicate (A,D,E,F) or duplicate (B,C). G and H are both representatives of two independent experiments. BFA, brefeldin A; cfDNA, cell-free DNA; DC, dendritic cell; moDC, monocyte-derived dendritic cell; Mφ, macrophage; PBMC, peripheral blood mononuclear cells; RPMI, Roswell Park Memorial Institute 1640 Medium.
As with the recombinant DNASE1L3 protein, the 206Cys variant impeded secretion of endogenous DNASE1L3 from moDCs of healthy humans homozygous for the rs35677470 risk allele by an average of 10-fold (figure 3B). Blocking secretion of DNASE1L3 led to increased intracellular protein in all genotype groups (figure 3C). Nucleosomal fragmentation of endogenous cell-free DNA from moDCs and in plasma, as well as exogenous nuclear chromatin by plasma, was significantly reduced (but not absent) in homozygous carriers of the 206Cys genotype, compared with 206Arg homozygotes (figure 3D–H). Equimolar comparisons of DNASE1L3 enzyme activity by genotype were not possible because DNASE1L3 protein levels in plasma were below the detection limits of the ELISA and western blot assays. Secretion of DNASE1L3 by heterozygous moDCs was much less than the intermediate amount of protein secretion expected, and enzyme activity was not reduced in association with protein levels.
Discussion
Here we showed that conditioning on the coding polymorphism rs35677470 eliminates the SLE risk association signal in the DNASE1L3/PXK locus. The lead SNPs rs180977001 and rs73081554 tag the same SLE risk haplotype as rs35677470. A secondary signal conferring SLE protection remained following conditioning on rs35677470. The most residual associated SNP is rs11130643, which shares a haplotype with a previously reported SLE protective haplotype,17 a haplotype which we also propose to be protective for RA and scleroderma (online supplemental table S4). The DNASE1L3 locus was previously associated with SLE in GWAS12 14; however, these studies did not report the rs35677470 missense variant association due to the absence of rs35677470 and any of its informative proxies on the SNP arrays used in those studies. Consequently, rs35677470 was missed altogether, or poorly imputed (ie, imputation score of 0.8112), resulting in loss of significant risk association. The functional basis of the protective haplotype remains to be established.
The Arg206Cys variant was originally reported to eliminate DNASE1L3 enzyme activity.15 However, a complete lack of enzyme activity is inconsistent with the effect size of disease risk in human subjects homozygous for 206Cys, given that DNASE1L3 deficiency leads to a monogenic form of lupus involving anti-DNA autoimmunity (usually presenting as hypocomplementemic urticarial vasculitis syndrome).4–7 We found that the Arg206Cys substitution reduces DNASE1L3 protein secretion, but that the limited amounts of secreted 206Cys protein still retain functional enzyme activity for the artificial and physiological substrates of plasmid and nuclear chromatin, respectively. Reduced DNASE1L3 enzyme secretion is consistent with reduced but not absent endonuclease activity reported by others.2 We also observed that nucleosomal fragmentation of cell-free DNA in plasma as well as enzyme activity in plasma correlated strongly to genotype, consistent with our in vitro observations. Given that circulating DNASE1L3 prevents the manifestation of anti-DNA autoimmunity in response to extracellular self-DNA, it follows that enzyme activity of DNASE1L3 206Cys in vivo is reduced due to a reduction in secreted protein, and subsequently, we expect that reduced clearance of extracellular self-DNA leads to increased risk of anti-chromatin autoimmunity (online supplemental figure S2).
Reizis and colleagues ascertained that the TLR signalling pathway (and not the STING pathway) is involved in the innate immune sensing of DNA via MyD88, initiating the anti-DNA immune response in Dnase1l3 knockout mice.2 20 Two potentially distinct sources of natural DNA antigen in the anti-DNA autoimmune response are apoptotic microparticles and neutrophil extracellular traps (NETs) derived from neutrophils undergoing NETosis. The unique C-terminal tail of DNASE1L3 confers its capacity for enzymatic activity on DNA in liposomes and enables DNASE1L3 endonuclease activity for chromatin DNA associated with NETs and microparticles.2 19 21 Microparticles are capable of eliciting an anti-DNA immune response in an animal model, and neutrophil NETs, in addition to being a candidate source for antigenic chromatin, are widely implicated in the pathogenesis of SLE. Of particular relevance, a high proportion of serum NET remnants correlated with low DNase enzyme activity in some patients with lupus nephritis.5 Importantly, DNase I and DNASE1L3 are necessary to clear neutrophil NETs and ensure survival in an animal model of sterile neutrophilia,21 implying that these DNases have evolved to prevent the effects of an overwhelming response by neutrophils during an immune response to infection. Further studies are needed to determine if Dnase1l3 knockout mice spontaneously develop anti-dsDNA antibodies to environmental triggers that favour the appearance of apoptotic microparticles or neutrophil NETs. Furthermore, the factors (genetic, tissue-specific or other) which drive the development of autoimmune disease characterised by anti-dsDNA antibodies versus anti-citrullinated protein antibodies or anti-centromere antibodies (present in rs35677470 associated SLE, RA8 or SSc9 11 respectively) also remain to be explored.
Given that rs35677470, rs180977001 and rs73081554 all tag the same risk haplotype, that loss-of-function variants in DNASE1L3 result in Mendelian forms of autoimmune disease, that rs35677470 is the categorical SNP associated with RA and SSc within this locus and that the missense variant confers a drastic secretion phenotype on the encoded DNASE1L3 protein, we conclude that rs35677470 is the causal variant for the association of the SLE risk haplotype at chromosome 3p14.3.
Data availability statement
Data are available upon reasonable request. The dataset analysed was from a publication by two of our authors (CDL, HCA, DSCG et al. Transancestral mapping and genetic load in systemic lupus erythematosus. Nat Commun 2017;8:16021. doi: 10.1038/ncomms16021 (published Online First: 2017/07/18)), and requests for SLE Immunochip data should be sent to CLangefe@wakehealth.edu.
Ethics statements
Ethics approval
The Genotype and Phenotype (GaP) Registry at The Feinstein Institutes for Medical Research provided fresh, de-identified human blood; blood was collected from consented control subjects under an institutional review board (IRB)-approved protocol (IRB# 09–081) and processed to isolate plasma and peripheral blood mononuclear cells. The Committee for Participant Protection at The Feinstein Institutes for Medical Research approved this study (TAP0307.5.85). The GaP is a subprotocol of the Tissue Donation Program at Northwell Health and a national resource for genotype–phenotype studies (https://www.feinsteininstitute.org/robert-sboas-center-for-genomics-and-human-genetics/gap-registry/). Subjects were selected on the basis of rs35677470 genotype from Immunochip and/or the Global Screening Array.
Acknowledgments
We thank Gila Klein, Kristine Elmaliki and Margaret DeFranco of the Genotype and Phenotype Registry, and the participants who provided blood, Lior Brimberg and Delcora Campbell for technical assistance, Betty Diamond and Sun Jung Kim for reagents and helpful discussion, and Michael Ryan, Bibu Jacob and staff members of the Boas Center Biorepository at the Feinstein Institutes for Medical Research for plasma and control genomic DNA.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
LNC and HW contributed equally.
Contributors KRS, LNC, MHG, CNM, AS and PKG conceived and designed the study; LNC and HW collected the experimental data; MC and CDL contributed genetics data; MC, CDL and WL performed the statistical genetics analysis; KRS, LNC and PKG wrote the paper. All authors read and approved the manuscript.
Funding This work is supported by a career development award to KRS (NIH K01AR071502).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.