Article Text

Abstract
For more than a decade, genome-wide association studies have been applied to autoimmune diseases and have expanded our understanding on the pathogeneses. Genetic risk factors associated with diseases and traits are essentially causative. However, elucidation of the biological mechanism of disease from genetic factors is challenging. In fact, it is difficult to identify the causal variant among multiple variants located on the same haplotype or linkage disequilibrium block and thus the responsible biological genes remain elusive. Recently, multiple studies have revealed that the majority of risk variants locate in the non-coding region of the genome and they are the most likely to regulate gene expression such as quantitative trait loci. Enhancer, promoter and long non-coding RNA appear to be the main target mechanisms of the risk variants. In this review, we discuss functional genetics to challenge these puzzles.
- arthritis
- rheumatoid
- autoimmune diseases
- immune complex diseases
- polymorphism
- genetic
Statistics from Altmetric.com
Introduction
Autoimmune diseases, such as rheumatoid arthritis, are thought to arise from the interaction of genetic and environmental factors. The contribution of genetic factors to autoimmune diseases has been examined for more than 40 years. Initial investigations focused on major histocompatibility complex genes and were followed by those that focused on several other candidate genes. Improvements in commercial arrays since 2007 has led to the development of the current form of genome-wide association studies (GWASs). GWASs have been applied in many studies investigating autoimmune diseases and have expanded our understanding of their underlying genetic factors. GWASs rely on single-nucleotide polymorphisms (SNPs) and haplotype blocks, which are distributed across all chromosomes. A number of representative tag SNPs—500 000 to several millions—are selected based on the haplotype information and combined into a single array for typing. The loci associated with disease susceptibility can then be determined by comparing variants represented by tag SNPs between patients and controls and identifying those that are significantly different.
GWASs have helped identify thousands of loci as being statistically associated with a risk for various diseases and traits.1 The same loci have also been identified in other independent analyses of the same diseases, suggesting that the associations determined using GWASs hold true.2 Genetic risk factors are essentially causative for specific diseases and traits; however, understanding the biological mechanism of risk from a genetic factor is challenging. Although there are only three possible patterns, with corresponding intermediate phenotypes, in which risk variants are involved in the development of a disease (figure 1), the types of change brought about by risk variants are complex. Furthermore, many variants significantly associated with each other are often in strong linkage disequilibrium (LD) and form a haplotype.3 Thus, although associations between variants and diseases can be identified, it is difficult to identify the causal variant among multiple variants located on the same haplotype,4 and experimental validations are needed to determine the functional variants.5 Furthermore, understanding the biological functions of risk variants is challenging, since more than 80% of disease-associated variants are located in non-coding regions of the genome.6 Transcriptome analysis can offer some clues regarding how these variants alter gene regulation and the expression of splice variants of target genes,7 but the detailed relationship between risk variants and genes is largely unknown.

Risk patterns of causative variants and intermediate phenotypes. Disease development involves a variety of characteristic intracellular and internal events. These are collectively referred to as intermediate phenotypes when we consider a causal genotype and the disease outcome. Intermediate phenotypes involve gene expression, protein expression and epigenetic effects and others. There are three possible patterns via which causative variants are associated with disease. First, a risk-associated genotype acts on the disease via the intermediate phenotype, which is assigned as the causal intermediate phenotype. Second, the intermediate phenotype is not causal but the consequence of the disease. Third, intermediate phenotypes are independent from the disease.
Analyses that integrate various ‘omics’ studies and GWAS data have emerged as powerful tools for understanding the functions of risk variants identified in a GWAS. The capabilities of next-generation sequencing techniques for analysing the functions of non-coding regions have advanced dramatically in recent years, enabling the comprehensive analysis of enhancers, promoters, histone modifications and chromatin structures. Analysis of the expression of quantitative trait loci (QTL) is used to investigate how particular variants lead to intermediate phenotypes (eg, epigenetic statuses). QTL are DNA markers on a chromosome that indicate genes involved in a quantitative trait.8 Recently, QTL analysis has been used to identify loci that are associated with a particular quantitative phenotypic trait or disease and can be caused to polygenic effects.9 Therefore, QTL analysis has been used for assessing human polygenic diseases and phenotypes as well as intermediate phenotypes, such as gene expression.
In this review, we discuss the possible functional genomic strategies for integrating GWAS results with the current understanding of specific diseases. In particular, we focus on promoters, enhancers and long non-coding RNAs. We also list a suite of analytical methods for the functional dissection of disease-associated risk loci. We believe that functional genomics will enhance our understanding of the immunological and biological functions of genes.
Functions of GWAS-identified risk variants
The ever-increasing number of genome-wide genotyping arrays has enabled the imputation of variants and the compilation of larger and more accurate reference datasets.10–12 Identifying the function of risk variants is important for understanding the mechanism of disease onset. Indeed, several risk variants affect the structures of proteins, for example, by inducing amino acid alterations or splicing variations. However, the functions of the majority (more than 80%) of risk variants are unknown because many of them are located in non-coding genomic regions. To elucidate the functions of risk variants, more detailed GWAS and omics data regarding intermediate phenotypes of the whole genome are necessary.
Post-GWAS omics analyses can improve our understanding of the molecular events orchestrating the development of diseases and traits. In particular, high-throughput transcriptomic3 7 and genome-wide epigenomic3 analyses are commonly used omics analyses that can be exploited to identify QTL. QTL identified through omics analyses overlapping with GWAS-identified risk variants hint at the pathogenetic mechanism of a disease. There are various types of QTL, which we have described below. The most common QTL type is expression QTL (eQTL), which is defined as a variant directly related to the expression level.
Expression QTL
The majority of disease-associated variants reside in non-coding regions of the genome, suggesting that they regulate gene expression. Indeed, a combination of statistical analysis, imputed genotypes in LD, transcriptomics and epigenomics has helped identify a number of eQTL that overlap with variants implicated in autoimmune diseases.3 Since eQTL are strongly influenced by epigenetic changes, they are often observed in conjunction with epigenetic QTL (figure 2). In many cases, eQTL function as cis-eQTL, which directly affect the expression of nearby genes (usually within 250 kb to 1 Mb) in an allele-specific manner.7 13 14 In contrast, eQTL that are located at distance from their gene-of-origin, often on different chromosomes, are referred to as trans-eQTL. Some cis-eQTL associated to house-keeping genes, are observed in many different tissue types; however, the majority of eQTL are highly tissue specific.12 The 3D map of chromatin interaction can be used to contextualise eQTLs. Hi-C and other chromosome conformation capture technologies provide a quantitative and qualitative description of the 3D arrangement of the genome and further our understanding of gene regulation.15

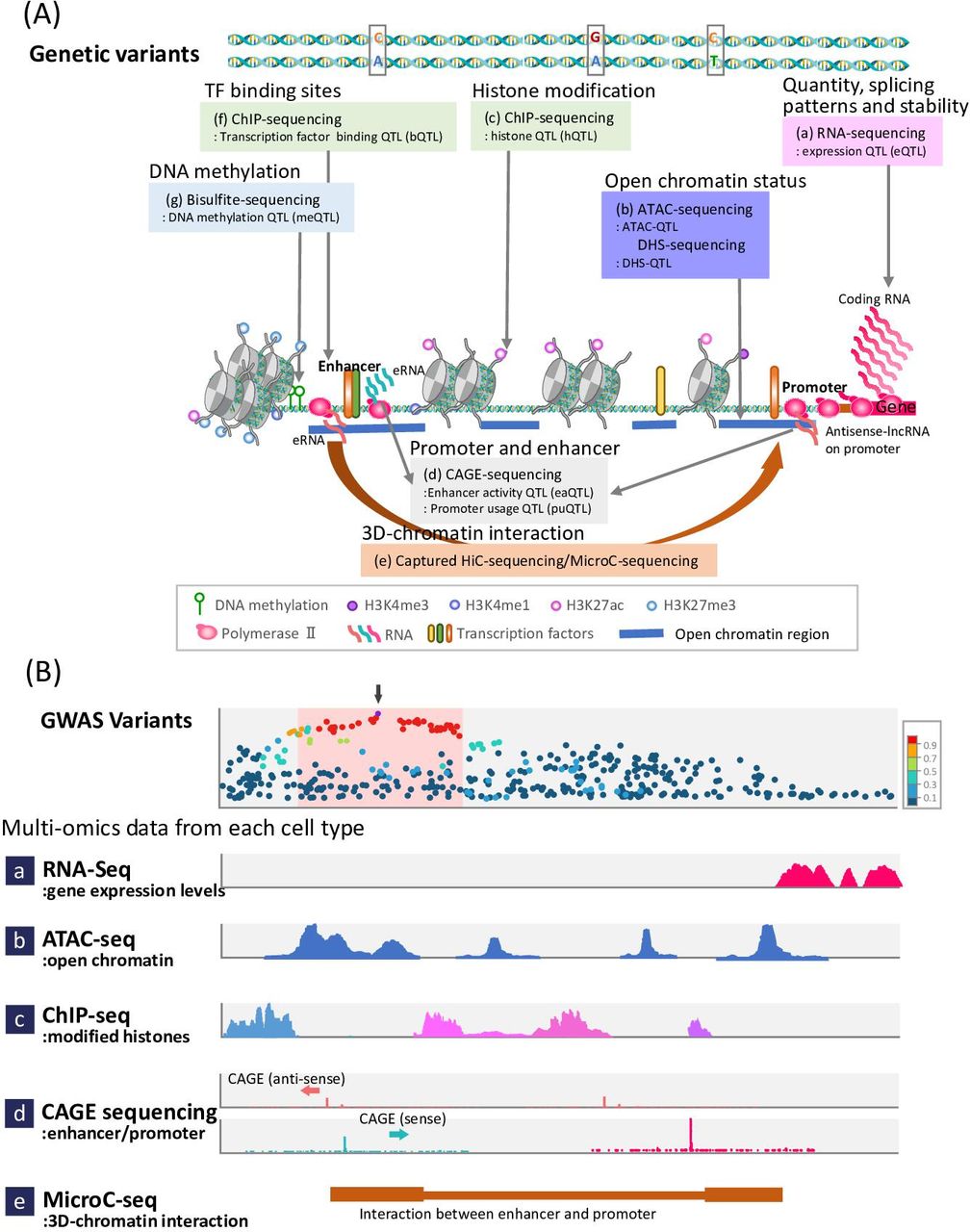
Genetic variants and quantitative trait loci (QTL). (A) Genetic variants can affect different types of transcriptional and post-transcriptional regulatory factors, including (a) gene expression level, (b) open chromatin status, (c) histone modifications, (d) promoter usage and enhancer activity, (e) chromatin conformation, (f) transcription factor binding sites, and (g) DNA methylation. (B) Actually, genome-wide association studies (GWASs) results integrate a variety of omics layer data constructed using the latest technologies (eg, the eQTL study involves RNA-seq for exploring gene expression profiles). In the upper row, the regional association plot of GWAS is presented. The purple dot indicates the top single-nucleotide polymorphism (SNP) (SNP with the smallest p value). SNPs highly correlated with the top SNP are equally likely to be causal in the LD block (pink box), demonstrating how omics data can be combined with GWAS findings to provide insights into causal variants and genes. These SNPs are usually outside the gene body (pink peaks, a) and are located in the regulatory region of the gene. The region is open (assay for transposase-accessible chromatin (ATAC)-peaks (blue), b) and flanked by modified histones (light blue and pink peaks, c). The non-coding RNA-expressed region (d) indicates active promoter and enhancer positions, which interact (Micro-C, brown bars, e) with each other. Integration of these data for each cell provides evidence for genes and cell types implicated in the disease GWAS findings. CAGE, cap analysis of gene expression; ChIP, chromatin immunoprecipitation.
Splicing QTL
Splicing QTL (sQTL), a subtype of eQTL, enable the quantitation of genetic variation due to the expression of RNA isoforms from alternative splicing events. sQTL are thought to contribute significantly to disease development because splicing is a major post-transcriptional modification and the primary mechanism governing protein diversity. Alternative splicing affects multiexonic genes, often in a cell type-specific fashion, and modulates phenotypic changes in a flexible and dynamic manner.16 RNA-sequencing (RNA-seq) is used for analysing both eQTL and sQTL.16 In multiple sclerosis, the estimated effect size of sQTL is larger than that of eQTL, supporting the well-known importance of RNA splicing in the brain.3 Thus, RNA splicing is a promising area of investigation to understand the effect of genetic variation in a complex disease.
Epigenetic QTL, including DNA methylation QTL and histone modification QTL
Epigenetic events are classically defined as post-translational modifications that determine chromatin status, including DNA methylation and multiple types of histone modifications (eg, H3K4me1, H3K4me3 and H3K27ac; figure 2). DNA methylation QTL (meQTL) and histone modification QTL are directly affected by their different genotypes. Epigenetic QTL are cell type and context specific.17 Indeed, epigenetic QTLs are highly specific for disease conditions, such as malignancy, or the effects of specific drugs. It is tempting to speculate that the high degree of specificity inherent to epigenetic QTL can be exploited to identify novel patient-specific therapeutic targets.
Assay for transposase-accessible chromatin QTL and DNase I hypersensitive site QTL
Gene expression and epigenetic events involve chromatin status, which is affected by both genetic variants and environmental factors. DNase I hypersensitive sites (DHS) indicate an open chromatin structure influence and are influenced by transcriptional regulatory elements and histone modifications (figure 2).9 Assay for transposase-accessible chromatin sequencing (ATAC-seq) also detects open chromatin. Genetic variants that are correlated with the phenotypes of DHS and ATAC are called DHS-QTL and ATAC-QTL, respectively. These QTL contribute to differences in gene regulation between individuals.18
Transcription factor-binding site QTL
Sequence-specific transcription factors (TFs) regulate gene expression by binding to cis-regulatory elements in the promoter and enhancer regions. TFs typically recognise binding-site motifs with particular DNA sequences (figure 2). Variants in DNA TF binding sites are designated as TF-binding site QTL (bQTL). For example, the binding site of transcription factor 21 has been reported to have bQTL effects.19
Promoter usage QTL and enhancer activity QTL
eQTL variants are typically enriched in regulatory regions. In particular, variants located in promoter regions are associated with alternative transcript usage and include variations in the mRNA 5’-end. These are termed promoter usage QTL (puQTL; figure 2).18 On average, there are more than four promoters per human gene, indicating that studies of puQTL are important for understanding eQTL effects.18
Enhancers are located remotely from promoters, and, unlike promoters, they regulate gene expression in a distance-independent manner by impacting transcriptional timing and cell-specificity of gene expression. Variants related to enhancer activity are referred to as enhancer activity QTL (eaQTL; figure 2). Methods that capture the 5'-end of a transcript are often used to identify puQTL and eaQTL.20
Non-coding RNA QTL
Micro RNAs (miRNAs) and long non-coding RNAs (lncRNAs) represent a class of non-coding RNAs that act as post-transcriptional regulators of gene expression and mRNA translation.21 miRNAs and lncRNAs are transcripts composed of 21–25 bp and >200 bp, respectively, and are extensively reported to be involved in transcriptional regulation (figure 1). We describe the relationship between promoters, enhancers and long non-coding RNAs in detail below.
Promoters and risk variants
Promoters are regulatory modules located upstream of the transcription start site (TSS). They are approximately 100–1000 bp long and contain a core region and one or more proximal regions.22 The core promoter supports the assembly of the transcription preinitiation complex, including general TFs and RNA polymerase (Pol) II.23 Core promoters control the magnitude of transcription, but not its induction, by regulating the binding of RNA Pol II at the TSS. Once the RNA Pol II reaches a few bases downstream of the TSS, further transcript elongation requires the binding of TFs and cofactors to the proximal region of the promoter.24–26
Genetic variants located in promoter regions, originally designated as eQTL, influence the strength and/or stability of the interaction between TFs and the promoter of an immune system-related gene, likely modifying its expression pattern and thus facilitating the onset of autoimmune diseases.27 For example, a variant altering the binding affinity for the transcription factor C/EBPβ in the promoter of FasL increases the risk of developing systemic lupus erythematosus.28 Similarly, an eQTL variant in the promoter of FCRL3, highly expressed in B cells, increases its affinity for NF-κB binding, rendering it a risk factor for rheumatoid arthritis.29
Promoters are widespread; thus, promoter risk variants that alter gene expression cannot always be determined by eQTL analysis but are revealed by puQTL analysis. Cap analysis of gene expression (CAGE), which detects the 5’-end of target transcripts, can help detect puQTL and eaQTL.20 30 In some cases, the results of CAGE are not linked to eQTL results. For example, TTC23 has two promoter regions 1.6 kb apart; rs8028374 was mapped as a puQTL with significant opposite effects on the two promoter regions. However, TTC23 has not been identified as an eQTL.18
Risk variants are found to be enriched in regulatory elements and are therefore thought to regulate gene expression levels, often in a cell type- and context-specific manner (eg, stimulation and disease conditions). Overall, the relative participation of alternative promoters in the transcriptional output of genes, a previously underappreciated factor affecting disease development and complex trait associations, but the integration of puQTL and eQTL in various cell types and cell conditions provides new insights into the mechanisms underlying eQTL and helps explain the prevalence of specific transcript isoforms.18
Enhancers and risk variants
Enhancers are distal regulatory modules that boost the transcription of associated promoters, irrespective of their orientation. The first enhancer was identified in vitro decades ago as a distal sequence that increased the transcription of a reporter gene under the control of the SV40 promoter.24 Enhancers typically span 50–1500 bp and contain multiple TF binding sites, enabling them to orchestrate proper spatiotemporal gene expression during development31 or in response to signalling molecules.32 Enhancers are structurally and functionally similar to the proximal regions of promoters. Indeed, both promoters and enhancers are bound by multiple TFs, support the formation of the preinitiation complex, and recruit RNA Pol II.33 Functionally, both promoters and enhancers support bidirectional transcription and activate transcript elongation.33 In promoters, the sense strand expresses mRNA and the antisense strand expresses promoter upstream transcripts (PROMPT/uaRNA). In enhancers, both the sense strand and the antisense strand express non-coding enhancer RNAs (eRNA) which are short, typically unspliced and non-adenylated transcripts.
Most GWAS variants have been found to overlap with cell type-specific enhancers that have been newly annotated through epigenomic profiling (table 1).34 35 Enhancer variants probably play an important role in fine-tuning the transcriptional output of each cell type and therefore affect susceptibility to common diseases. A number of classical methods to identify enhancers rely on the evolutionary conservation of non-coding sequences.35 36 In a landmark study, the candidate causal variants for 21 autoimmune diseases were integrated with RNA-seq data and chromatin features of control and stimulated human CD4+ T-cell subsets, regulatory T cells, CD8+ T cells, B cells and monocytes.3 Importantly, 90% of the causal variants were located in the non-coding regions of the genome. Approximately 60% of the variants were mapped to enhancers specific to immune cells, which showed increased histone acetylation and transcriptional activity on immune stimulation.3 The same study demonstrated that causal disease variants had a high probability of being context-specific immune enhancers and suggested that most non-coding causal variants alter atypical regulatory motifs rather than recognisable consensus motifs.3 Validation of enhancers and promoters identified through GWASs has historically been highly challenging.
Genome-wide association studies (GWASs)-associated expression quantitative trait loci (eQTL) affecting gene regulatory elements
More recently, next-generation sequencing technologies used in large-scale epigenomics projects have enabled the mapping and characterisation of regulatory regions in the human genome. Several methods have been developed to efficiently detect enhancers, such as chromatin immunoprecipitation-sequencing (ChIP-seq), which is used to identify TF binding or a histone modification in the enhancer sequence, and the DHS-seq and ATAC-seq methods, which are used to identify the open chromatin structure of activated enhancers. These methods were initially used in combination to detect enhancers.37 As direct detection methods of regulatory regions, self-transcribing active regulatory region sequencing (STARR-seq) and CAGE-sequencing have been developed, which are used for assaying the enhancer activity of millions of enhancer candidates from arbitrary sources of DNA.38 In combination with next-generation sequencing, CAGE-sequencing enables the genome-wide determination of coding and non-coding TSSs.20 33 39 The resulting database is instrumental to the identification of promoters and enhancers, both of which are characterised by the bidirectional transcripts. Enhancer activation leads to the bidirectional production of low levels of eRNAs.40
Effects of lncRNAs and other transcripts from non-coding regions
The cumulative expression of non-coding genes is approximately four times greater than the expression of all mRNAs, making lncRNAs one of the most abundant RNA species within the average cell.9 The genomic and epigenomic characterisation of lncRNA-coding regions revealed that lncRNAs preferentially originate from enhancers rather than promoters.20 lncRNAs do not encode proteins; however, lncRNAs longer than 200 nucleotides fold into complex structures and have biological functions. Functional lncRNAs can specifically interact with proteins, DNA and other RNAs to exert their molecular functions (figure 3). Folded lncRNAs have a variety of functions, such as transcriptional regulation (figure 3A), recruitment of TFs and proteins (figure 3B), degradation of transcript and splicing variants (figure 3C), and chromatin remodelling (figure 3D).20 41 42

{kind=link}
{kind=link}
{kind=link}
Genetic variants and long non-coding RNAs (lncRNAs). Genetic variants can affect the quantification and function of lncRNAs. (A) lncR-QTL, (B) regulation of transcription by lncRNAs, (C) binding of RNAs and lncRNAs, resulting in the formation of alternative splicing isoforms, and (D) interaction of lncRNAs with chromatin-modifying enzymes and nucleosome-remodelling factor. QTL, quantitative trait loci.
lncRNAs are a key component of the repertoire of regulatory elements that control cell differentiation and maintain tissue homeostasis. Recently, lncRNAs have been clearly implicated in immune cell lineage commitment and immune responses (table 2). They have also emerged as an important class of molecules implicated in various human cancers as well as cardiovascular, neurodegenerative, and autoimmune diseases.43 44 If variants with eQTL effects are located on lncRNAs, they may affect transcriptional regulators.20 Therefore, the relationship between lncRNAs and risk variants in diseases and traits has received a significant amount of attention and should be the focus of future research.
Examples of lncRNAs involved in immune cell functions
Genome-wide lncR-eQTL analysis is expected to uncover how GWAS variants relate with lncRNA expression in common diseases and phenotypes. A comprehensive eQTL analysis was previously performed using genome-wide lncRNA expression and genotype data from human peripheral blood mononuclear cells of 43 unrelated individuals. Subsequently, various omics integrative network analyses were applied to construct variant-lncRNA-mRNA interaction networks, resulting in the detection of 707 pairs of cis-effect associations (p<5.64E−06) and 6657 trans-effect associations (p<3.51E−08).45
The integration of genetic data sets from 1829 expression profiles in the FANTOM5 project uncovered the existence of nearly 20 000 functional lncRNAs. Approximately 2000 lncRNAs were expressed in significantly associated tissue/trait pairs. For example, the association between the middle temporal gyrus and autism spectrum disorder involves 18 lncRNAs from 49 genes.20 On the other hand, a genetic variant over-represented in patients with coeliac disease lies within lnc-13, reduces its binding affinity for HDAC1, and weakens the epigenetic repression of inflammatory genes.46 lncRNAs regulate multiple processes in disparate subsets of immune cells, such as the intensity of STAT3 signalling in human dendritic cells,47 Th1 versus Th2 differentiation of human lymphocytes,42 and perhaps not surprisingly, the epigenetics-mediated training of immune responses in human monocytes.41 Thus, lncRNAs play an important role in gene regulation, and their profiling can reveal associations with specific tissues and cells.
Further analyses for functional genomics
Several different analyses have been used to annotate putative causal variants in GWASs, such as risk-associated SNPs in the promoter and enhancer regions (figure 2B; table 1).48 Some of the functions of risk-associated enhancers and promoter variants have been experimentally proven, while others have been inferred from positional information; however, the functions of enhancers are particularly difficult to prove. In fact, the functions of many risk variants that exist outside of gene bodies remain unclear. Enhancers may be far away from the TSS, even further than 1 Mb. Transcriptional regulation from distal locations is facilitated by the folding of the genome, which places regulatory elements in three-dimensional proximity of the TSS. Advances in analytical techniques and next-generation sequencing techniques, like Hi-C-sequencing and micro-C-sequencing, allow the identification of trans-acting regulatory elements as well as cis -acting elements49–51 (figure 2B). Integrated analysis of omics data is also currently undergoing various statistical advances.
Biological analysis of regions regulated by TFs that were previously analysed by reporter assays can now be comprehensively performed through massive parallel reporter assays (MPRAs). In MPRAs, a library of barcoded candidate regulatory sequences is generated, cloned in the backbone of a reporter plasmid, and then transfected into an appropriate cell line. RNA-seq then allows the precise identification and quantitation of the transcriptional output associated with each regulatory variant. MPRAs facilitates the high-resolution, quantitative dissection of sequence-activity relationships of transcriptional regulatory elements with high reproducibility. MPRAs were developed to test the function of enhancer sequences in combination with targeted mutagenesis52 53 and have subsequently been exploited to test human promoters, enhancers54 and eQTL.55 56 However, episomal reporter plasmids do not allow the testing of candidate regulatory sequences in their original context, a limitation that may affect the validation of epigenetic QTL. Chromatinised adenoviral and lentiviral vectors have therefore been developed to circumvent this limitation.57 58
A new type of screening assay based on CRISPR/Cas9 technology examines the functions of regulatory regions in their original genomic context. CRISPR/Cas9 is used to test the function of candidate regulatory regions by targeted genomic deletion.59 Other CRISPR-based screening approaches, such as CRISPR/Cas9–mediated activation and inactivation, have been coupled with RNA-seq-based transcriptional quantification. However, these methods have not yet been scaled to the genome-wide level.60 Furthermore, new approaches that combine CRISPR-based genome engineering with patient-specific induced pluripotent stem cell-based models are powerful tools for identifying putative causal loci that regulate gene expression and cellular functions.61
Advances in next-generation sequencing have improved single-cell gene expression analysis. Bulk RNA-seq experiments, commonly used in transcriptomic studies, examine the average gene expression among sampled cells and thus mask cell heterogeneity. However, single-cell RNA sequencing (scRNA-seq) can be used to reveal the heterogeneity of gene expression between various cells. Large-scale scRNA-seq can reveal patterns of gene, isoform, and allelic expression across different cell types and cell conditions.62 Most scRNA-seq methods use unique molecular identifiers (UMIs) of specific allelic origin.62 Utilisation of UMIs confers an important advantage to eQTL analysis, since they are useful to precisely understand allele imbalance. However, scRNA-seq analysis methods have low detection sensitivity and may not be able to detect many genes with low levels of expression, suggesting a need for improvement.63 New statistical analyses have been developed to enable cell type identification, such as deconvolution analysis, which uses scRNA-seq and reference panel data. In addition to the transcriptomic analysis, scRNA-seq is also used in epigenomic analyses (eg, single-cell ChIP-sequencing). Additionally, spatial transcriptomics methods are also improving markedly.64 In the future, spatial transcriptomics methods may enable the visualisation, while retaining the spatial information of the tissue.
Conclusions
As described above, considerable progress has been made in understanding the genetics and pathogenesis of autoimmune diseases. However, our understanding of the causality of these diseases is inadequate, and the development of new therapies based on genomic information for these diseases is required. Therefore, we need to apply a variety of approaches to identify the missing links.
GWASs are based on the hypothesis that commonly occurring diseases involve commonly occurring variants, with a large number of variants being involved in the development of a disease. Accordingly, functional analyses of risk variants are complicated. Statistical associations between genes and diseases alone cannot identify the risk variants that contribute to pathogenesis. Integration of GWAS data and omics data has the potential to elucidate the function of each risk variant while also identifying disease and tissue specificity. These findings will help reveal a more detailed mechanism of pathogenesis of autoimmune diseases and thus lead to their precise diagnosis and treatment.
Ethics statements
Ethics approval
Our study is performed in accordance with the International Conference on Harmonisation Guidelines for Good Clinical Practice and the Declaration of Helsinki, and the study protocols were approved by ethics review boards of RIKEN.
References
Footnotes
Handling editor Josef S Smolen
Contributors AS and MMG carried out the experiment. AS wrote the manuscript with support from KY and MMG. KY supervised the project.
Funding This study was funded by Japan Society for the Promotion of Science (grant number 18H05285) and Chugai Pharmaceutical Co.
Competing interests KY has received grants from Chugai Pharmaceutical Co.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.