Article Text

Abstract
Objective Systemic lupus erythematosus (SLE), an autoimmune disorder, has been associated with nearly 100 susceptibility loci. Nevertheless, these loci only partially explain SLE heritability and their putative causal variants are rarely prioritised, which make challenging to elucidate disease biology. To detect new SLE loci and causal variants, we performed the largest genome-wide meta-analysis for SLE in East Asian populations.
Methods We newly genotyped 10 029 SLE cases and 180 167 controls and subsequently meta-analysed them jointly with 3348 SLE cases and 14 826 controls from published studies in East Asians. We further applied a Bayesian statistical approach to localise the putative causal variants for SLE associations.
Results We identified 113 genetic regions including 46 novel loci at genome-wide significance (p<5×10−8). Conditional analysis detected 233 association signals within these loci, which suggest widespread allelic heterogeneity. We detected genome-wide associations at six new missense variants. Bayesian statistical fine-mapping analysis prioritised the putative causal variants to a small set of variants (95% credible set size ≤10) for 28 association signals. We identified 110 putative causal variants with posterior probabilities ≥0.1 for 57 SLE loci, among which we prioritised 10 most likely putative causal variants (posterior probability ≥0.8). Linkage disequilibrium score regression detected genetic correlations for SLE with albumin/globulin ratio (rg=−0.242) and non-albumin protein (rg=0.238).
Conclusion This study reiterates the power of large-scale genome-wide meta-analysis for novel genetic discovery. These findings shed light on genetic and biological understandings of SLE.
- lupus erythematosus
- systemic
- polymorphism
- genetic
- epidemiology
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. The meta-analysis summary association statistics in the current study are available from the corresponding author on reasonable request.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Genome-wide association studies have identified nearly 100 susceptibility loci for systemic lupus erythematosus (SLE) risk.
The known SLE loci explain partially the disease heritability.
What does this study add?
This study identified 113 genomic regions including 46 novel loci for SLE risk.
The study prioritised 110 putative causal variants including 10 putative causal variants with high confidence (posterior probability ≥0.8).
How might this impact on clinical practice or future developments?
These findings revealed new genetic basis for SLE and generated molecular mechanisms hypotheses for further investigations.
Introduction
Systemic lupus erythematosus (SLE) is an autoimmune disorder characterised by the production of autoantibodies that damage multiple organs.1 Considerable genetic predisposition contributes to SLE aetiology.2 To date, nearly 100 susceptibility loci have been identified for SLE, mainly through genome-wide association studies (GWASs).3–8 However, these loci collectively only explain ~30% of SLE heritability9 and their biology, in terms of causal variants, effector genes and cell types and pathological pathways that mediate genetic effects, has not yet been fully characterised.10
Genome-wide association meta-analyses have been performed to uncover new genetic associations for SLE in Asians,11 Europeans12 and trans-ancestral populations.9 However, the study sample sizes were relatively modest, which limits their ability for genetic discovery. GWASs have successfully linked genetic variants with human common diseases and traits.13 Nonetheless, only ~8% of GWAS participants are East Asians.14 East Asians have a unique population genetic history and may have ethnicity-specific genetic architecture involved in the development of disease and manifestations. For example, SLE has a remarkably higher prevalence and younger age of onset in Asians.15 16 Genetic heterogeneity may explain, at least partly, the phenotypic diversity of SLE between East Asians and Europeans.9 Hence, large-scale East Asian investigations may provide an opportunity to identify unique genetic associations even for the same diseases and traits that have already been well studied in Europeans.17
Methods
Study participants
We recruited a total of 10 029 SLE cases and 180 167 healthy controls in three independent case–control cohorts from mainland China, Korea and Japan. We analysed additionally 3348 SLE cases and 14 826 controls that were published in our previous East Asian SLE GWASs4 6–9 to increase statistical power. All the cases fulfilled the revised American College of Rheumatology SLE classification criteria or were diagnosed by collagen disease physicians (online supplemental table 1). Each participant provided written informed consent.
Supplemental material
Genome-wide association analyses
We newly genotyped 10 029 SLE cases and 180 167 controls, and revisited raw genome-wide genotype data in 3348 SLE cases and 14 826 controls from the five published studies.4 6–9 Quality controls were conducted for each of the eight data sets. Genotype imputation was accomplished using reference panels from the 1000 Genomes Project (1KGP) phase 3 v518 and population-specific reference panels19 in IMPUTE2/420 21 or MINIMAC4.22
We tested association between SLE risk and genotype dosages in each data set using a logistic regression or linear mixed model in PLINK,23 SNPTEST24 or EPACTS (https://genome.sph.umich.edu/wiki/EPACTS) (online supplemental table 1). Within each data set, we filtered out association results based on imputation quality (IMPUTE info or MINIMAC r2 ≤0.3), minor allele frequency (MAF) ≤0.5% or Hardy-Weinberg equilibrium test p<1.0×10−6 in controls. For each cohort, the association analysis for the X chromosome was conducted separately by sex and then meta-analysed across both men and women. For data sets analysed using a linear mixed model (online supplemental table 1), allelic effects and standard errors were converted to a log-odds scale to correct for case–control imbalance.25
Fixed-effects meta-analysis
We aggregated the association summary statistics from the eight data sets using a fixed-effects inverse-variance meta-analysis in METAL.26 We applied a genomic control correction to each association summary statistic. Heterogeneity in allelic effect sizes among data sets was assessed using Cochran’s Q statistic. We excluded genetic variants available in only a single data set. We defined SLE susceptibility loci by merging ±250 kilobases (kb) windows around genome-wide associated variants to ensure that lead single nucleotide polymorphisms (SNPs) were at least 500 kb apart. We defined lead variants as the most significant SLE-associated variant within each locus. A locus was considered novel if the lead SNP was at least 500 kb away from any previously reported SLE-associated variants.
Approximate conditional association analysis
To dissect distinct association signals at each SLE locus, we performed an approximate conditional analysis using GCTA COJO27 with genome-wide meta-analysis summary statistics based on linkage disequilibrium (LD) estimated from 7021 unrelated Chinese controls. The Chinese reference individuals for LD calculation were retrieved from the Chinese study using the Illumina Infinium Global Screening Array data (online supplemental table 1), excluding first-degree and second-degree relatives.
Bayesian statistical fine-mapping analysis
To prioritise causal variants in SLE susceptibility loci, a statistical fine-mapping analysis was performed using FINEMAP v1.4 software,28 with meta-analysis z-scores and LD matrices estimated from the 7021 Chinese reference individuals. We used default priors and parameters in FINEMAP, assuming at most five causal signals in the ±250 kb region around a lead variant at each SLE locus. FINEMAP computed a posterior probability (PP) for each genetic variant being the true putative causal variant. For each association signal, we ranked the candidate putative causal variants in a descending order of their PPs, and then built a 95% credible set of causal variants by including the ordered variants until their cumulative PP reached 0.95.
Heritability estimation by LD score regression
Overall SLE heritability h2 explained by genome-wide variants was estimated using the LD score regression model29 with LD scores18 from the 1KGP East Asian descendants, based on an SLE population prevalence of 0.03% in East Asian populations.1 SLE heritability estimate was further partitioned according to known and novel SLE loci using stratified LD score regression.30 The boundary of each SLE locus was arbitrarily defined as ±500 kb flanking the lead SLE-risk variant.
Genetic correlation between SLE and other traits by LD score regression
We calculated genetic correlations between 98 traits (39 diseases17 and 59 quantitative traits31 and SLE by using bivariate LD score regression.32 We used the LD scores18 from the 1KGP East Asian descendants, limited the genetic variants to the HapMap3 SNPs and removed the variants with extended human leucocyte antigen (HLA) region (chromosome 6: 25 to 34 megabases (Mb)).
Patient and public involvement
Patients and the public were not involved in the design or analysis of this study.
Results
Identification of 46 novel SLE susceptibility loci
We performed a large genome-wide association meta-analysis in 13 377 SLE cases and 194 993 controls of East Asians (online supplemental table 1). To the best of our knowledge, this is the largest genetic association study of SLE to date. The effective sample size (Neff=50 072) is three-fold and four-fold larger than that of the largest published trans-ancestry9 and East Asian11 meta-analyses, respectively.
We tested associations for 11 270 530 genetic variants in a fixed-effects meta-analysis. A quantile–quantile plot showed that test statistics were well-calibrated, with a genomic-control inflation factor λGC=1.06 (indicating that ancestry effects had been well controlled; online supplemental figure 1). LD score regression29 showed that polygenic effects (89.4%), rather than biasses, primarily caused the inflation residual (estimated mean χ2=1.32 and LD-score intercept=1.03).
We detected 26 379 genetic variants associated with SLE at p<5×10−8 within 113 loci (figure 1A and online supplemental table 2), of which 46 were novel (table 1). The pairwise LD between lead variants was low (LD r2 <0.002). For seven novel loci, MAFs of the lead SNPs were 10-fold higher in East Asians than in Europeans (figure 1B). Two of them and their LD neighbours (r2 ≥0.2 in either East Asians or Europeans) would be undetectable in Europeans with the same effective sample size and risk magnitude due to low statistical power (<10%; online supplemental table 3).

Summary of meta-analysis association results and comparison of MAFs for lead variants within the 46 novel loci between East Asians and Europeans. (A) Manhattan plot of genome-wide association meta-analysis results from 208 370 SLE East Asians including 13 377 SLE cases and 194 993 controls. Minus log10-transformed association p values (y-axis) are plotted along chromosomal positions (x-axis). Known and novel loci are highlighted in light blue and pink, respectively. The red dashed line denotes the genome-wide association significance threshold of p=5×10−8. The grey dashed line represents p=10−30, at which the y-axis breaks. (B) Comparison of MAFs of lead variants within the 46 novel loci between East Asians (y-axis) and non-Finnish Europeans (x-axis) in the Genome Aggregation Database (gnomAD) v3. Variants with more than 10 times higher MAFs in East Asians are coloured purple above a red dashed line. MAF, minor allele frequency.
Association results for the 46 novel susceptibility loci for systemic lupus erythematosus
Associations at exonic variants
The meta-analysis identified lead missense variants in two novel loci (CHD23 and LRRK1; figure 2A,B and online supplemental table 2). In addition, we detected three new exonic variants (including two missense variants) within the reported SLE loci including CSK (rs11553760), IKBKB (rs2272736) and TYK2 (rs55882956) genes (figure 2C–E and online supplemental table 2). They were not correlated with previously reported exonic variants within the same genes (LD r2 <0.02 in East Asians or Europeans; online supplemental table 4), suggesting possible allelic heterogeneity of these genes. We replicated four known associations for missense variants at AHNAK2 (rs2819426),33 IRAK1 (rs1059702),34 NCF2 (rs13306575) and WDFY4 (rs7097397; online supplemental table 2).35 36

New lead exonic variants identified at three known (CSK, IKBKB and TYK2) and two novel (CHD23 and LRRK1) loci. (A) rs11553760 (synonymous variant) at CSK. (B) rs2272736 (p.Arg303Gln, missense variant) at IKBKB. (C) rs55882956 (p.Arg703Trp, missense variant) at TYK2. (D) rs10823829 (synonymous variant) at CHD23. (E) rs35985016 (p.Lys203Glu, missense variant) at LRRK1. The lead SNP is labelled as purple diamond. The LD is estimated from 7021 Chinese samples. LD, linkage disequilibrium; Mb, megabases; SNP, single nucleotide polymorphism.
Secondary association signals within SLE loci
To dissect the source of association signals at each locus, we conducted an approximate conditional analysis using GCTA27 with meta-analysis summary statistics and LD estimates from 7021 unrelated Chinese controls. We acknowledge the limitations of using LD estimation from a single population for a meta-analysis of diverse East Asians. We identified a total of 233 independent association signals with conditional p<5×10−8, 169 of which arose from non-HLA regions (online supplemental table 5). We observed from two to four signals at each of 28 non-HLA loci (including seven novel loci). For example, we discovered two distinct association signals within the known STAT4 locus, including the previously reported SNP rs1188934112 and the new insert-deletion variant (indel) rs71403211 (figure 3A). For the 46 novel loci, we discovered 55 distinct signals (online supplemental table 5 and figure 2). We noticed that most of the signal index variants (n=190, 82%) are common (MAF ≥5%) with modest effects (online supplemental table 5).

Two independent association signals identified. (A) At two intronic variants within known STAT4 locus. (B) At known (rs7097397, p.Arg1816Gln) and new (rs7072606, p.Ser214Pro) missense variants within WDFY4 locus. (C) A known intronic variant within ATXN2 gene and a new (rs1131476, p.Ala352Thr) missense variant within OAS1 gene. The lead and secondary index variants are labelled in diamond. The lead variant and its LD proxies are in red while the secondary signal index variant and its LD proxies are in blue. The LD is estimated from 7021 Chinese samples. LD, linkage disequilibrium; Mb, megabases.
Approximate conditional analysis detected two novel missense variants at WDFY4 and OAS1 genes. We detected two distinct signals within WDFY4, including the known (rs7097397)37 and a new (rs7072606) missense variant (LD r2=0.02 between two variants in East Asians), which suggests allelic heterogeneity at this locus (figure 3B). We provided for the first time genome-wide association evidence at a missense variant within OAS1 (rs1131476, LD r2=0.78 with rs1051042, which is a known missense variant but only exhibited suggestive significance with SLE in previous study,33 figure 3C and online supplemental table 5).
Prioritisation of causal variants
To prioritise putative causal variants, we conducted a Bayesian statistical fine-mapping analysis for 111 loci using FINEMAP28 after excluding complex associations involving the HLA and 7q11.23. We found exactly the same number of association signals in 57 loci between FINEMAP causal configuration with the highest posterior probability and the GCTA approximate conditional test. To be conservative, we only summarised the statistical fine-mapping results for these 57 regions, which contained 65 association signals (online supplemental table 6).
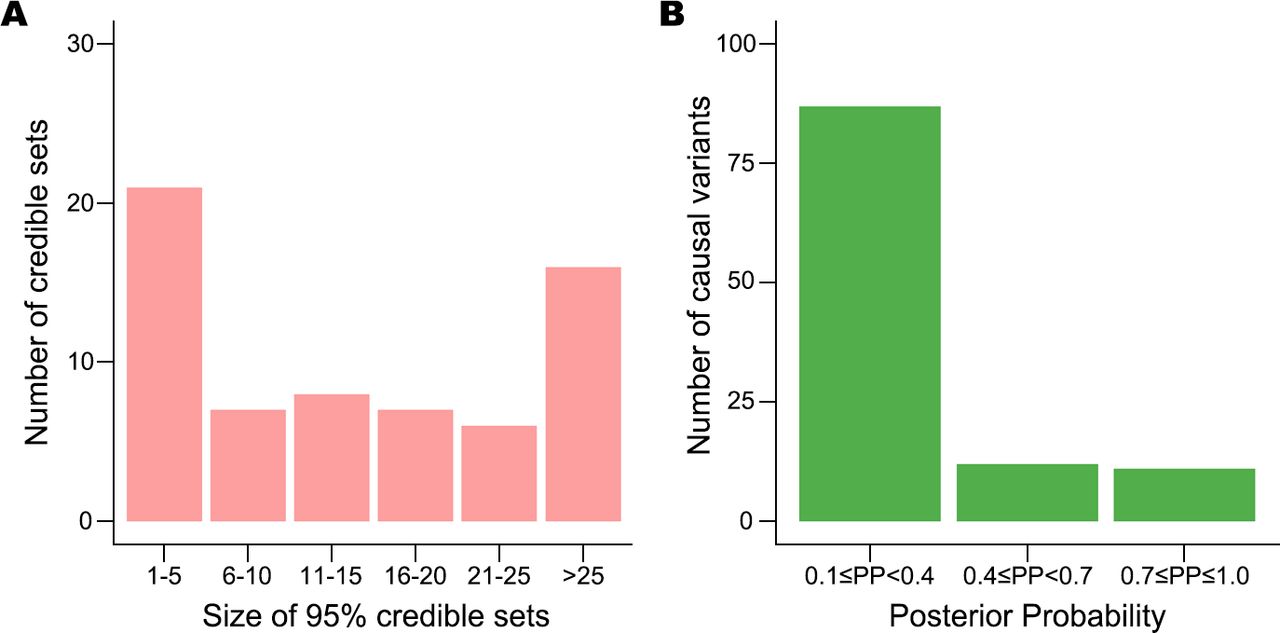
For each signal, we built a credible set of putative causal variants with a 95% probability of including the true causal variants. The size of 28 credible sets was small (size ≤10; figure 4A). Among the 110 putative causal variants with posterior probability ≥0.1 (figure 4B), we found four coding variants (3.6%), which implies that most of these associations are probably induced by non-coding causal variants. The prioritised variants are available to be tested as potential targets in perturbation experiments. For example, the allele-specific regulatory activity of the intronic variant (rs10036748) with the highest posterior probability (0.387) in the TNIP1 locus was recently experimentally characterised in SLE.38
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Results of statistical fine-mapping analysis. (A) Number of 95% credible sets of putative causal variants, binned by their sizes. (B) Number of potential causal variants with posterior probabilities (PP) ≥0.1, which are considered to be the true causal variants.
We pinpointed a single most likely causal variant with high confidence (posterior probability ≥0.8) for four known (ATXN2, BACH2, DRAM1/WASHC3 and NCF2) and six novel (17p13.1, ELF3, GTF2H1, LRRK1, LOC102724596/PHB and STIM1) loci (online supplemental table 6). For example, we prioritised rs61759532 as a putative causal variant at the novel 17p13.1 locus (PP=0.999). This variant is located in an intron of ACAP1, which encodes a key regulator of integrin traffic for cell adhesion and migration.39
SNP-based heritability
To assess the proportion of phenotypic variance explained by common variants, we applied LD score regression29 to the meta-analysis results. Assuming a population prevalence of 0.03% for SLE,1 we estimated the liability-scale SNP-based heritability from all non-HLA variants as h2 SNP = 7.24% (SE=0.78%). The 66 known and 46 novel non-HLA loci explained 62.6% (SE=4.9%) and 22.1% (SE=2.6%) of this overall SNP-based heritability, respectively.
Genetic correlation with other diseases/traits
To explore shared genetics between SLE and various traits, we calculated genetic correlations of SLE with 39 complex diseases and 59 quantitative traits in Biobank Japan participants using bivariate LD score regression32 (online supplemental table 7). As expected, we detected significant positive genetic correlations between SLE and two other autoimmune diseases: rheumatoid arthritis (rg=0.437) and Graves’ disease (rg=0.318). In addition, we found unreported genetic correlations (FDR<0.05) with albumin/globulin ratio (rg=−0.242) and non-albumin protein (rg=0.238).
Discussion
Here, we carried out the largest-ever genome-wide association meta-analysis for SLE and identified 113 risk loci including 46 novel regions for SLE in 208 370 East Asians including 13 377 SLE cases and 194 993 controls. This study revealed new genetic predispositions for SLE and generated hypotheses for further studies to investigate diseases functional mechanisms.
Epidemiological studies have found the higher prevalence of SLE in East Asians and heterogeneous disease manifestations across ethnicities.15 16 Previous investigations suggested genetics might explain the phenotypic heterogeneity.9 We observed that the MAFs of the index variants for several novel genetic associations were much higher in East Asians than in Europeans. Specifically, we suggested two novel loci were more likely specific to East Asians. These findings might help explain the genetic basis of SLE phenotypic heterogeneity between East Asians and Europeans. The results reinforce the power of large-scale genetic association for genetic discovery of SLE in relatively less studied populations.
We identified 11 exonic variants including two missense variants within novel loci CHD23 and LRRK1, four novel missense variants within known SLE loci IKBKB,9 TYK2,9 WDFY4 37 and OAS1, 33 and three known missense variants within known AHNAK2,33 IRAK1 34 and NCF2.35 36 These findings suggested allelic heterogeneity within several of these loci and highlighted the disease-risk effects of genes AHNAK2, CSK, IKBKB, IRAK1, NCF2, OAS1, TYK2 and WDFY4 within eight known loci, and CDH23 and LRRK1 within two novel loci which potentially alter gene product activity in an allele-specific manner. The novel gene CHD23 plays a role in cell migration40 while LRRK1 encodes a multiple-domain leucine-rich repeat kinase. A previous study observed that LRRK1-deficient mice exhibited a profound defect in B-cell proliferation and survival and impaired B-cell receptor-mediated NF-κB activation,41 which suggested that the association within this region might confer the risk of SLE through modulating the NF-κB pathway and the activities of B cells. We noted that the Bayesian statistical fine-mapping analysis prioritised the lead missense variant rs35985016 as the most likely putative causal variant for this association. This variant is highly frequent in our study individuals but is rare in Europeans. The molecular mechanisms in SLE risk worthy further investigations.
In the present study, we localised the putative causal variants for SLE genetic association in high resolution. Our findings indicated that the putative causal variants for the majority of SLE associations were non-coding variants. We provided targets of candidate putative causal variants with high confidence for several SLE loci. These findings are worthy for further exploration in functional experiments. We showed the regulatory effect of one of the putative causal variants in an accompanied paper. We acknowledged the limitation of a small LD reference panel from single population in the Bayesian statistical fine-mapping analysis.
We found for the first time the significant genetic correlations between SLE, albumin/globulin ratio and non-albumin protein. These findings might reflect the renal complications commonly developed in SLE patients who have been reported to have significantly lower albumin/globulin ratio and higher serum globulin than healthy controls in epidemiological studies.42 These shared genetic basis findings might suggest a common pathway underlying the SLE risk and kidney function in addition to the direct damage of SLE autoantibodies on kidney.
In summary, we detected 46 novel loci for SLE risk in the largest meta-analysis and prioritised putative causal variants for 65 causal signals. This study highlights the power of large-scale genetic association study in East Asian populations. The findings reveal the genetic predispositions for SLE and provide clues for further the investigation of disease mechanisms.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. The meta-analysis summary association statistics in the current study are available from the corresponding author on reasonable request.
Ethics statements
Ethics approval
The study protocol was approved by the Institutional Review Board at each participating institute and the meta-analysis study was additionally approved by the Institutional Review Boards at Anhui Medical University, Hanyang University Hospital of Rheumatic Diseases, and RIKEN Center for Medical Sciences.
Acknowledgments
We acknowledged the participants in this study. We appreciate the contribution of Japanese Research Committee on Idiopathic Osteonecrosis of the Femoral Head. We appreciate all contributors to BioBank Japan. Details are included in supplementary material.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
XY, KKim and HS contributed equally.
Contributors XY, KKim and HS contributed equally to this work, and either has the right to list himself first in bibliographical documents. SCB, YC, CT, XZhang, XY, KKim and HS conceived the study design. SCB, YC, XZhang, SY, KKim and CT acquainted the financial support. XY, KKim, HS, CT, YC and SCB wrote the manuscript. XY, KKim, HS, EH, XZheng, VL and YW conducted all of the analyses with the help of JBH, LCK, MTW, SP, SE, HS, KT, NO, MK, KI and C Terao. KKim, SYB, LW, LL, RXL, YSheng, MYH, WL, KYoon, MC, HH, MW, YTang, HD, CL, CS, WF, KL, BJK, HSL, SCB, SH, YSakamoto, NSugano, MM, DT, KKarino, TMiyamura, JN, GM, TKuroda, HN, TMiyamoto, TT, YKawaguchi, KA, YTada, KYamaji, MS, TA, AS, TSumida, YOkada, KMatsuda, KMatsuo, YKochi, TSeki, YTanaka, TKubo, RH, TYoshioka, MY, TKabata, YA, YOhta, TO, YN, AK, YY, KOhzono, KYamamoto, KOhmura, TYamamoto and SI generated genetic data. SYB, SJ, YCK, WTC, SSL, SCS, YMK, DY, CHS, YBP, JYC, YP, GYA, JMS, YKL, DJP, WY, THK, SY, BJK, NShen, HSL, XZhang, CT and SCB managed the cohort data. All authors reviewed and approved the manuscript.
Funding This research was supported by General Program (81872516, 81573033, 81872527, 81830019, 81421001), Young Program (81803117, 82003328), Exchange Program (81881340424), and Science Fund for Creative Research Groups (31630021) of National Natural Science Foundation of China (NSFC), Distinguished Young Scholar of Provincial Natural Science Foundation of Anhui (1808085J08), National Program on Key Basic Research Project of China (973 Program) (2014CB541901), China National Key R&D Program (2016YFC0906100), Science Foundation of Ministry of Education of China (213018A), Program for New Century Excellent Talents in University of Ministry of Education of China (NCET-12-0600), The Bio & Medical Technology Development Program of the National Research Foundation, funded by the Ministry of Science & ICT of the Republic of Korea (NRF-2017M3A9B4050355 to SCB), Basic Science Research Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT and Future Planning (2015R1C1A1A02036527 and 2017R1E1A1A01076388 to KKim), National BioBank of Korea, the Centers for Disease Control and Prevention, Republic of Korea (KBN-2018-031 to SSL), Center for Genome Science, Korea National Institute of Health, Republic of Korea (4845-301, 3000-3031 to MYH, KYoon and BJK), Japan Agency for Medical Research and Development (AMED) and the BioBank Japan project supported by the Ministry of Education, Culture, Sports, Sciences and Technology of the Japanese Government and AMED under grant numbers (17km0305002 and 18km0605001), Grant of Japan Orthopaedics and Traumatology Research Foundation, lnc, (No. 350 to YSakamoto), RIKEN Junior Research Associate Program (to H.S.), US NIH grants (AI024717, AI130830, AI148276, HG172111 and AR070549 to JBH), US Department of Veterans Affairs (BX001834 to JBH) and Center for Pediatric Genomics Award and CCRF Endowed Scholar Award of Cincinnati Children’s Hospital (to MTW).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.