Article Text

Abstract
Objective To compare the efficacy and safety of brodalumab, an interleukin-17 receptor subunit A inhibitor, with placebo, in patients with psoriatic arthritis (PsA).
Methods Adult patients with active PsA and inadequate response to, or intolerance to, conventional treatment were enrolled into two phase III studies (NCT02029495 and NCT02024646) and randomised 1:1:1 to receive subcutaneous brodalumab 140 mg or 210 mg or placebo at weeks 0, 1 and every 2 weeks up to 24 weeks. About 30% of patients had prior use of biologics. The primary endpoint for both studies was the American College of Rheumatology 20 (ACR20) response at week 16.
Results 962 patients were randomised across the studies prior to early termination due to sponsor decision. The primary endpoint was met in both studies. Based on comparable design and eligibility criteria, data from both studies were pooled. Significantly more patients achieved ACR20 at week 16 in both brodalumab treatment groups (45.8% and 47.9% for 140 mg and 210 mg, respectively) versus placebo (20.9%) (p<0.0001). Similar results were observed at week 24. Significantly higher proportions of patients receiving brodalumab achieved ACR50/70, Psoriasis Area and Severity Index 75/90/100 and resolution of dactylitis and enthesitis versus placebo (p<0.01). Adverse event rates were similar across treatments at week 16 (54.4%, 51.6% and 54.5% for placebo, brodalumab 140 mg and 210 mg, respectively). No new safety signals were reported.
Conclusion Brodalumab was associated with rapid and significant improvements in signs and symptoms of PsA versus placebo. Brodalumab was well tolerated, with a safety profile consistent with other interleukin-17 inhibitors.
- psoriatic arthritis
- autoimmune diseases
- DMARDs (biologic)
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Brodalumab has demonstrated efficacy in a phase II trial of patients with psoriatic arthritis (PsA).
What does this study add?
These phase III trials summarise the efficacy and safety of brodalumab in a much larger population, namely 962 patients with PsA.
How might this impact on clinical practice or future developments?
Receptor-level targeting of the interleukin-17 cytokine family involved in the pathogenesis of PsA by brodalumab results in clinically meaningful improvements in articular, enthesitis, dactylitis, skin and health-related domains. These trials provide important information for clinicians treating patients with PsA with brodalumab.
Introduction
Psoriatic arthritis (PsA) is a chronic inflammatory disorder that can affect the joints, tendon sheaths, entheses and axial skeleton.1 2 PsA is a heterogeneous condition with different clinical phenotypes, varying in severity, disease course and numbers of affected joints.3 Patients with PsA can experience substantial disability, with severe joint damage, digital deformation, functional impairment and impairment of quality of life (QoL).4
Current treatment guidelines recommend biologic disease-modifying antirheumatic drugs (DMARDs) as a treatment option on inadequate response following treatment with non-steroidal anti-inflammatory drugs (NSAIDs), corticosteroids and conventional synthetic DMARDs.5 6 Despite the advent of therapeutics targeting tumour necrosis factor (TNF), interleukin (IL)-17A and IL-12/23,5–7 and, more recently, Janus kinase and phosphodiesterase type 4, an unmet need remains in PsA as a significant proportion of patients either do not respond or eventually lose response to currently available therapies.5 6 8 Brodalumab is a fully human monoclonal antibody with a unique mechanism of action that binds to the IL-17 receptor subunit A (IL-17RA) with high affinity and, as a consequence, blocks the action of multiple proinflammatory cytokines of the IL-17 family, beyond that of IL-17A alone. Brodalumab 210 mg is currently approved for the treatment of moderate-to-severe plaque psoriasis9 10 in the USA, EU, Canada and certain Asian countries and for PsA currently only in Japan.11 The efficacy and safety of brodalumab in PsA were evaluated in a phase II, randomised, double-blind, placebo-controlled trial (NCT 01516957).12 Brodalumab 140 mg and 280 mg once every 2 weeks (Q2W) were associated with significantly greater improvements in clinical response (American College of Rheumatology 20 (ACR20); primary endpoint) versus placebo at 12 weeks. The safety profile of brodalumab in PsA was consistent with the safety profile established in the psoriasis clinical trial programme,13 14 and clinical responses were sustained during an open-label extension up to week 52.12
To further evaluate the efficacy and safety of brodalumab in PsA, two double-blind, randomised, phase III trials, AMVISION-1 (NCT02029495) and AMVISION-2 (NCT02024646), were conducted. The primary objective of both trials was to compare the efficacy of brodalumab with placebo in patients with PsA. Both trials were placebo controlled through week 24. Data at week 16 from individual trials and week 24 from a pooled analysis are presented.
Methods
Trial design and participants
AMVISION-1 and AMVISION-2 were multicentre, randomised, double-blind, placebo-controlled trials with planned long-term extensions. Both trials evaluated the efficacy and safety of subcutaneous brodalumab at doses of 140 mg and 210 mg Q2W in patients with active PsA who had an inadequate response or intolerance to conventional treatment with NSAIDs and/or DMARDs. The trial protocols were approved by an independent ethics committee or institutional review board at each trial site, and the trials were conducted in accordance with the International Conference on Harmonisation guideline on Good Clinical Practice. All patients provided written informed consent at trial procedure commencement.
Patients were aged ≥18 years with a diagnosis of PsA for ≥6 months at the time of enrolment and fulfilled the Classification for Psoriatic Arthritis criteria2 at screening. Full inclusion and exclusion criteria are presented in online supplemental methods. Briefly, patients were included if they had ≥3 tender and ≥3 swollen joints (excluding the distal interphalangeal joints of the feet as part of a 66/68 joint count), an active psoriatic skin lesion and a history of intolerance or inadequate response to NSAIDs and/or DMARDs. Concomitant NSAIDs, DMARDs (methotrexate, sulfasalazine or leflunomide) and corticosteroids (≤10 mg/day prednisone or equivalent) were permitted, provided the dose was stable for ≥4 weeks prior to initiation of trial treatment. Patients in AMVISION-1 had ≥1 erosion of the hands or feet or C reactive protein (CRP) ≥1.0 mg/dL. Patients were excluded if they had an active or history of infection (including active tuberculosis), Crohn’s disease, TNF inhibitor therapy within 2 months prior or other biologic therapy within 3 months prior to trial initiation or anti-IL-17 or anti-IL-12/IL-23 biologic therapy at any time. Patients with a prior history of suicidal ideation and behaviour (SIB) were excluded after the implementation of a protocol amendment. This amendment was implemented following the identification of SIB as a potential risk and after discussion with regulatory agencies. Specific tools were added to assess eligibility and monitor subject safety (ie, stopping rules). Three patients with a history of SIB were recruited into the studies prior to the amendment taking effect and continued in the trial after the amendment.
Supplemental material
The first patient was enrolled in AMVISION-1 on 6 March 2014 and in AMVISION-2 on 24 March 2014. Both trials were terminated on 24 June 2015.
Treatment and randomisation
Following a 4-week screening period, patients in each trial were randomised 1:1:1 to receive subcutaneous brodalumab 140 mg or 210 mg or placebo on day 1 and weeks 1, 2, then Q2W through week 22 (in online supplemental figure 1), stratified by baseline body weight, prior biologic use and geographical region using a permuted block design within each stratum. Biologic-experienced and biologic-naïve populations were each capped at no more than 60% of the global trial population; however, the final trial population included about 70% of biologic-naïve patients. This deviation is not considered to have an impact on the interpretation of the data. From week 14, patients were evaluated for inadequate response, defined as failure to achieve ≥10% improvement from baseline in tender and swollen joint counts at two consecutive scheduled visits where joint counts were assessed (eg, weeks 14 and 16). If the criteria for inadequate response were met, initiation and/or dose adjustments of non-biologic treatments were permitted. Patients on placebo with inadequate response were switched to brodalumab 210 mg, and from week 24, patients who were originally randomised to placebo and had not already met criteria for inadequate response received brodalumab 210 mg with an additional dose at week 25. From week 28 through week 34, patients who did not achieve ≥10% improvement from baseline in their tender and swollen joint counts at any visit, despite ≥12 weeks of continuous treatment after meeting inadequate response criteria, were considered non-responders and treatment was discontinued. Further information on inadequate response, rescue treatment and other criteria for permanent discontinuation is provided in the online supplemental appendix. Both trials were planned with a 52-week double-blind treatment phase, followed by a long-term open-label extension phase. After treatment assignments were unblinded, all patients subsequently received open-label brodalumab at their current Q2W dose.
Endpoint
The primary endpoint for both trials was ACR20 response at week 16. Secondary efficacy endpoints included ACR 50/70 and Psoriasis Area and Severity Index (PASI) 75/90/100 response rates; change from baseline in Health Assessment Questionnaire-Disability Index (HAQ-DI); Disease Activity Score with a 28-joint count and CRP; improvement in dactylitis assessed as present (yes or no) on 20 digits and enthesitis assessed as present (yes or no) on six entheses); Psoriatic Arthritis Disease Activity Score (PASDAS); and Clinical Disease Activity Index score (CDAI). A radiographic endpoint (evaluated through modified Total Sharp Score) was included in AMVISION-1. Due to the premature cessation of the trial and the subsequent smaller trial population recruitment, no definitive conclusions could be drawn with respect to the radiographic progression endpoint; as such, the data are not reported herein. Disease Activity in Psoriatic Arthritis (DAPSA) was included as a post hoc analysis. The primary and all secondary endpoints were also evaluated at week 24. The safety profile of brodalumab was evaluated in all patients who had received ≥1 dose of trial drug by recording adverse events (AEs), serious AEs (SAEs), laboratory assessments and vital signs. Safety at week 16 was reported as number and percentage of subjects who reported AEs, and at week 24 as number of AEs and AE rate/100 patient years, due to the early termination of the studies.
Statistical analyses
The trials were designed to detect significant treatment differences in ACR20 response between the brodalumab and placebo arms at week 16 with >90% power, assuming the underlying rate of response was 45% and 18% in the brodalumab and placebo arms, respectively.
Both studies were terminated by the sponsor (Amgen) prior to reaching their recruitment targets. The power of the primary endpoint in both studies, although reduced, was still sufficient. To account for all randomised patients and those who did not have the opportunity to complete the trial, non-responder imputation (NRI) and a generalised estimating equation (GEE) model were implemented. The GEE model included prior biologic use, geographical region, baseline body weight and treatment by visit interaction terms as fixed effects. Under the assumption that missing data were missing completely at random (MCAR) the estimated treatment effects derived from the GEE model would be unbiased. For this analysis, patients qualifying for rescue treatment or withdrawing from the trial for any reason other than trial termination were treated as non-responders. Results of the GEE analysis are presented in this manuscript by trial and as a pooled analysis. Further information on trial termination, analysis of continuous endpoints and subsequent statistical analysis is provided in the online supplemental methods.
Results
Patients
At the time of trial termination, in AMVISION-1 478 of the planned 630 patients, and in AMVISION-2, 484 of the planned 495 patients had been enrolled. The majority of these patients (693, 72%) completed 24 weeks of treatment. Patient disposition is summarised in online supplemental figure 2. The main reasons for discontinuation during the first 24 weeks were sponsor decision (19.8%) and withdrawal of consent (7.6%).
Demographics and baseline disease characteristics were balanced across treatment groups in both trials (table 1). The mean age across the trials was approximately 48 years, 50% of patients were women, and at baseline, one-third of patients had received prior biologic treatment in all treatment groups. Approximately two-thirds of patients had psoriasis covering ≥3% of their body surface area. Mean swollen and tender joint counts were 12 and 21, respectively. Fifty per cent of the patients had dactylitis (score >0) and nearly 67% of patients had enthesitis (score >0); 30% of patients had prior use of biologics.
Patient demographics and baseline disease characteristics (all randomised patients)
Efficacy
The primary objective was met, with significantly greater proportions of patients achieving ACR20 with both brodalumab doses versus placebo at week 16 (table 2 and figure 1A). Pooled analysis of data from both trials showed ACR20 response rates of 45.8% and 47.9% for brodalumab 140 mg and 210 mg, respectively, versus 20.9% for placebo at week 16 (both p<0.0001 vs placebo; table 2 and figure 1A). The marginal response rate was maintained through 24 weeks (51.0%, 53.6% and 23.8% for brodalumab 140 mg, 210 mg and placebo, respectively; both p<0.0001 vs placebo).

(A) ACR20, (B) ACR50 and (C) ACR70 response rates from baseline to week 24. Full analysis set. Dashed line represents the primary endpoint for each study. Response rates (95% CI) were calculated using a GEE model. NRI was applied following early withdrawal from trial for reasons other than premature study termination, and to subjects who satisfied the inadequate response criteria prior to week 24. GEE analysis assumed missing data due to early trial termination and intermittent missing data were MCAR. ACR responses were modified based on 66/68 joint counts. ACR20/50/70, American College of Rheumatology 20/50/70% improvement criteria; GEE, generalised estimating equation; MCAR, missing completely at random; NRI, non-responder imputation.
Comparison of brodalumab versus placebo at weeks 16 and 24 using generalised estimating equation
Summary of safety: adverse events up to week 16 (safety population, pooled analysis)
Other endpoints were also significantly improved in brodalumab recipients. Higher ACR50 response rates were observed for brodalumab versus placebo at all time points from week 2 for brodalumab 210 mg and week 4 for brodalumab 140 mg (figure 1B), and higher ACR70 response rates were observed versus placebo from week 8 for brodalumab 210 mg and week 12 for brodalumab 140 mg (figure 1C). Generally, the proportions of patients achieving ACR responses were higher in the brodalumab 210 mg group versus the 140 mg group.
A significantly higher proportion of patients with dactylitis at baseline achieved resolution in both brodalumab groups at weeks 12, 16 and 24 versus placebo (table 2 and figure 2A), with higher observed rates of dactylitis resolution with brodalumab 210 mg versus 140 mg at these time points. Among patients with enthesitis at baseline, resolution was achieved by a significantly higher proportion in both brodalumab groups versus placebo at weeks 16 and 24 (table 2 and figure 2B). Furthermore, brodalumab treatment resulted in significantly greater mean changes in HAQ-DI, CDAI, DAPSA and PASDAS scores from baseline versus placebo at weeks 16 and 24. Finally, the proportions of patients achieving a minimally important difference in HAQ-DI scores from baseline (defined as 0.3515) were significantly higher with brodalumab versus placebo at weeks 16 and 24 (table 2).
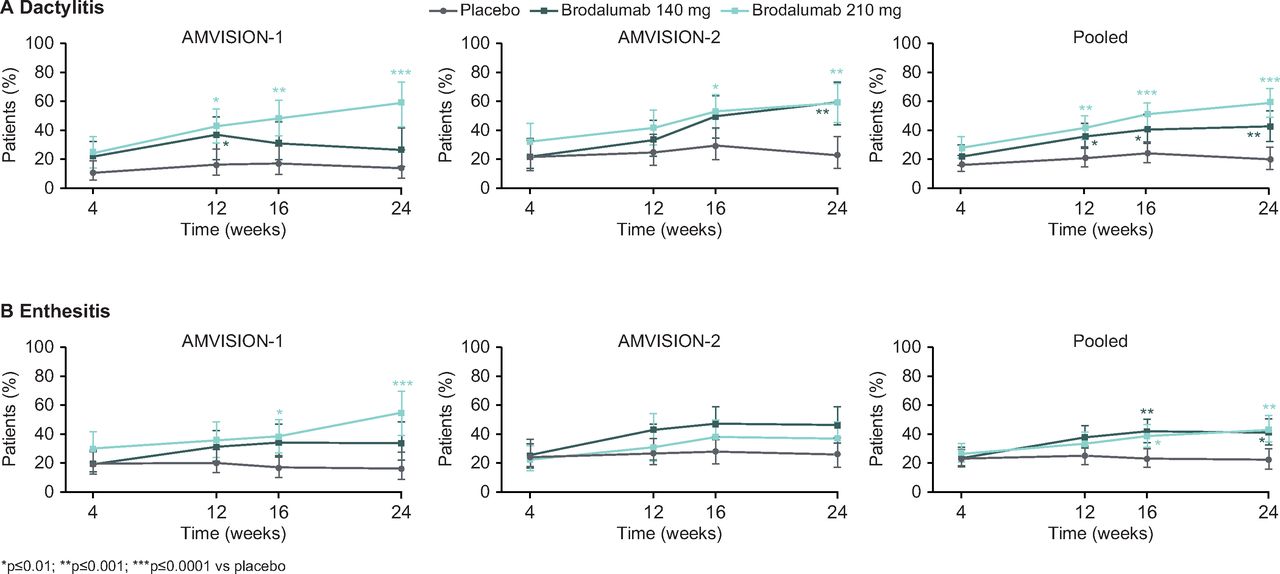
Resolution of dactylitis and enthesitis. Full analysis set. Response rates (95% CI) were calculated using a GEE model. NRI was applied following early withdrawal from trial for reasons other than premature trial termination, and to subjects who satisfied the inadequate response criteria prior to week 24. GEE analysis assumed missing data due to early trial termination and intermittent missing data were MCAR. Dactylitis and enthesitis responses were evaluated in patients with these conditions at baseline. Dactylitis was assessed as present (yes/no) on 20 digits (fingers and toes). Enthesitis was assessed as present (yes/no) on six entheses (lateral epicondyle, medial femoral condyle and Achilles’ tendon insertion). GEE, generalised estimating equation; MCAR, missing completely at random; NRI, non-responder imputation.
Brodalumab also demonstrated efficacy in skin-related endpoints. A significantly higher proportion of patients in both brodalumab groups achieved PASI75 versus placebo at all time points from week 2 (table 2 and figure 3A). Similarly, significantly more patients achieved PASI90 (figure 3B) and PASI100 (figure 3C) at all time points from week 4 onwards. There was a clear dose-dependent difference over time in the proportion of patients achieving PASI75/90/100 in the brodalumab 140 mg and 210 mg groups, with greater proportions of patients achieving PASI75/90/100 at the higher brodalumab dose.

{kind=link}
{kind=link}
{kind=link}
(A) PASI75, (B) PASI90 and (C) PASI100 response rates from baseline to week 24. PASI responses were calculated using the psoriasis efficacy full analysis set (patients with baseline BSA ≥3%). Response rates (95% CI) were calculated using a GEE model. NRI imputation was applied following early withdrawal from trial for reasons other than premature trial termination, and to subjects who satisfied the inadequate response criteria prior to week 24. GEE analysis assumed missing data due to early trial termination and intermittent missing data were MCAR. BSA, body surface area; GEE, generalised estimating equation; MCAR, missing completely at random; NRI, non-responder imputation; PASI75/90/100, 75/90/100% improvement in Psoriasis Area and Severity Index.
Safety
In the pooled analysis, the total duration of exposure to trial treatment was 107.8, 121.7 and 123.9 patient years in the placebo, brodalumab 140 mg and 210 mg groups, respectively. Overall, the percentage and type of AEs reported in the brodalumab 210 mg group at week 16 was similar to that of placebo and most events were mild to moderate in severity (table 3). The proportion of patients reporting AEs of interest was low (most commonly infection (24% to 30%)). No major imbalances or emergent safety signals were detected. Safety data at week 24 (event rates per 100 patient years) were similar to those at week 16 (online supplemental table 3). There were no deaths throughout the duration of the trials; the event rate of SAEs with brodalumab was low. Patients treated with brodalumab experienced a numerical increase in cases of neutropenia versus placebo, but none was related to infection. Serious infections were reported infrequently at week 24, with only one event recorded in the brodalumab 210 mg group in AMVISION-1 during the trial period (urosepsis reported during the first 16 weeks, which resolved). One patient with a history of SIB and depression in the brodalumab 140 mg group in AMVISION-2 was diagnosed with suicidal ideation following the first completed Columbia Suicide Severity Rating Scale 8 days after the first dose of brodalumab. The event was not considered related to trial medication by the investigator and resolved on the same day. The patient continued in the study for about another year without reporting other SIB events. Injection site reactions were generally mild and infrequent.
Discussion
The heterogeneity of PsA requires treatment options that are active across all disease domains (including skin, arthritis, dactylitis and enthesitis). The results from AMVISION-1 and AMVISION-2 demonstrate that brodalumab provided rapid and significant improvement compared with placebo in the signs and symptoms of PsA. The primary objective was met in both trials, and improvements were observed in articular, enthesitis, dactylitis, skin and QoL domains. These data suggest that brodalumab, with its unique mechanism of action, can offer clinical benefit to patients with PsA and thus reassure clinicians using brodalumab in people with psoriasis that musculoskeletal benefits can also accrue.
Brodalumab is a fully human monoclonal antibody that binds to the IL-17RA with high affinity. By blocking the IL-17RA, brodalumab inhibits the action of multiple proinflammatory IL-17 family cytokines (IL-17A, IL-17F, IL-17A/F, IL-17E (IL-25) and IL-17C).16–18 These cytokines all play broad roles in the type 17 T cell pathway, including complex crosstalk and endogenous control of the inflammatory response, and their dysregulation can lead to the destruction of tissue and the pathogenesis of autoimmune diseases such as psoriasis and PsA.19–31
Overall, the trial populations were representative of patients with PsA. Significant differences versus placebo were observed for endpoints related to joint involvement (ACR20/50/70) as well as those associated with skin manifestations (PASI75/90/100) and were accompanied by an improvement in patient-reported physical function (HAQ-DI). Throughout the trials, response and improvement in musculoskeletal and psoriasis endpoints were generally greater among patients who received the higher brodalumab dose. This trend was more marked in the AMVISION-1 trial. In order to enrich the trial population for evaluation of radiographic progression, the trial population of AMVISION-1 consisted of patients who had a more severe disease at baseline, as compared with patients in AMVISION-2. Consequently, a dose–response may have been more easily observed in AMVISION-1 than in AMVISION-2. The onset of effect occurred as early as 2 weeks after initiation of treatment with the 210 mg brodalumab dose for some endpoints such as PASI75. Specifically for AMVISION-2, nominal and statistical improvement in both brodalumab groups was evident despite the response rate in the placebo group (ACR20, 24.8%) being in the upper range of placebo response rates previously reported in other studies of biologics treating similar PsA populations (11% to 24% for IL-17, IL-12/-23 and TNF inhibitors).32–36
The trials were terminated early (24 June 2015) following a decision from the sponsor (Amgen) to stop its participation in the codevelopment of brodalumab after events of SIB had been observed in the clinical programme and an anticipation that it would lead to restrictive labelling, (refer to online supplement for further information). Given that the assessments of treatment effects at weeks 16 and 24 were both clinically relevant, as well as the trajectory of the response over the duration of treatment, the GEE model was chosen as it provided a more succinct presentation of the clinical trial data. In this model, missing data due to trial termination and intermittent missing data were assumed to be MCAR. Monotone missing data due to reasons other than early trial closure were imputed using NRI, which assumes that patients who discontinued early, due to an AE, lack of efficacy and so on, would have been non-responders, had they remained in the trial. In essence, MCAR assumes that the decision to stop the trial/the reason for missing data is unrelated to the individual subject's ability to respond to treatment. The results of these analyses are robust regarding the statistical method and assumptions regarding missing data as the primary analysis for patients completing the week 16 visit prior to study termination closely matched the GEE analysis at week 16 (online supplemental table 4). In addition, the primary and GEE analyses at week 24 were also closely matched. Post hoc sensitivity analyses of ACR20 at week 24 were performed using different models and different methods for imputation of missing data. These analyses are consistent with the results of the primary analysis and confirm the robustness of the conclusions despite the change in model and imputation method from the original statistical analysis plan and protocol. Sensitivity analysis II, using NRI for all missing data, corresponds to the analysis specified in the protocol (see online supplemental table 2).
The incidences of AEs and SAEs with brodalumab were consistent with the known safety profile of brodalumab previously reported in psoriasis and PsA,12–14 with no increased rate of AEs related to brodalumab versus placebo, and no evidence of dose-dependence. Patients with a prior history of SIB were excluded from these trials after the implementation of a protocol amendment, allowing for an assessment of whether new instances of SIB were encountered in the brodalumab-treated population. The overall frequencies of depression and SIB in these trials were similar for brodalumab- and placebo-treated subjects during the double-blind period, suggesting that brodalumab treatment did not increase the risk for depression and SIB among patients with no prior history.
In summary, the AMVISION-1 and AMVISION-2 trials showed that brodalumab 140 mg and 210 mg Q2W are associated with substantial improvements in both joint-related and skin-related endpoints versus placebo in patients with PsA, and these improvements are maintained through 24 weeks. The safety profile of brodalumab was consistent with that reported in previous trials in psoriasis and the phase II PsA trial.12 The favourable safety profile and efficacy data from these trials suggest that inhibition of IL-17RA with brodalumab, a unique mechanism of action, may represent an additional treatment strategy for patients with PsA.
Supplemental material
Supplemental material
Acknowledgments
Medical writing support was provided by Ian Eustace, BSc (Hons) and Robyn Foster, PhD from Adelphi Communications Ltd, Bollington, UK, funded by LEO Pharma.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Josef S Smolen
Correction notice This article has been corrected since it published Online First. Table 2 has been corrected.
Contributors The AMVISION-1 and AMVISION-2 trials were sponsored by Amgen and AstraZeneca. The efficacy analyses presented were performed by KR from LEO Pharma, and the safety analyses presented were performed by Amgen/AstraZeneca. KR had full access to all the data in these studies and takes responsibility for the integrity of the efficacy data and the accuracy of the data analyses. All authors participated in interpreting the results and writing the manuscript and approved the final version for submission. AstraZeneca was not involved in the decision to publish these analyses and did not participate in the preparation of this manuscript.
Funding This study was funded by LEO Pharma.
Competing interests PJM reports, outside the submitted work, grants and personal fees from AbbVie, Amgen, Bristol Myers Squibb, Celgene, Janssen, Eli Lilly, Novartis, SUN Pharma, UCB Pharma; personal fees from Genentech, Boehringer Ingelheim, Galapagos, Gilead and GlaxoSmithKline. PSH reports, outside the submitted work, grants, personal fees and non-financial support from AbbVie; grants from Amgen, Celgene, Janssen, MSD, Pfizer and UCB; and personal fees from Galapagos. KFH was an employee of LEO Pharma at the time this study was conducted. KFH also reports, outside the submitted work, grants from AbbVie, Celgene, LEO Pharma and Novartis; personal fees from AbbVie, CSL Behring, Eli Lilly, Janssen, LEO Pharma, Novartis and Pfizer. KR is an employee of LEO Pharma. IBM reports personal fees from LEO Pharma during the conduct of the study. IBM also reports, outside the submitted work, grants and personal fees from Celgene, Compugen and UCB; grants from AstraZeneca, Novartis and Roche; personal fees from AbbVie, Galvani, Eli Lilly and Pfizer.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Ethics approval The study was conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonisation Guidance for Good Clinical Practice. Independent institutional review board approvals were obtained, and all patients provided written informed consent in accordance with local requirements.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement All data relevant to the study are included in the article or uploaded as supplementary information.