Article Text

Abstract
Objectives Genomic Risk Scores (GRS) successfully demonstrated the ability of genetics to identify those individuals at high risk for complex traits including immune-mediated inflammatory diseases (IMIDs). We aimed to test the performance of GRS in the prediction of risk for systemic sclerosis (SSc) for the first time.
Methods Allelic effects were obtained from the largest SSc Genome-Wide Association Study (GWAS) to date (9 095 SSc and 17 584 healthy controls with European ancestry). The best-fitting GRS was identified under the additive model in an independent cohort that comprised 400 patients with SSc and 571 controls. Additionally, GRS for clinical subtypes (limited cutaneous SSc and diffuse cutaneous SSc) and serological subtypes (anti-topoisomerase positive (ATA+) and anti-centromere positive (ACA+)) were generated. We combined the estimated GRS with demographic and immunological parameters in a multivariate generalised linear model.
Results The best-fitting SSc GRS included 33 single nucleotide polymorphisms (SNPs) and discriminated between patients with SSc and controls (area under the receiver operating characteristic (ROC) curve (AUC)=0.673). Moreover, the GRS differentiated between SSc and other IMIDs, such as rheumatoid arthritis and Sjögren’s syndrome. Finally, the combination of GRS with age and immune cell counts significantly increased the performance of the model (AUC=0.787). While the SSc GRS was not able to discriminate between ATA+ and ACA+ patients (AUC<0.5), the serological subtype GRS, which was based on the allelic effects observed for the comparison between ACA+ and ATA+ patients, reached an AUC=0.693.
Conclusions GRS was successfully implemented in SSc. The model discriminated between patients with SSc and controls or other IMIDs, confirming the potential of GRS to support early and differential diagnosis for SSc.
- scleroderma
- systemic
- immune complex diseases
- autoimmune diseases
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Systemic sclerosis (SSc) is a complex immune-mediated disease (IMID) for which a Genomic Risk Score (GRS) has never been implemented.
What does this study add?
A SSc GRS discriminates between patients with SSc and healthy controls with a remarkable predictive value.
Clinical information, such as serologic subtype and immune cells counts, adds accuracy to the GRS model.
The SSc GRS is capable of discriminating between SSc and other IMIDs.
How might this impact clinical practice or future developments?
This SSc GRS is a promising tool to improve the diagnosis and prognosis of patients with SSc.
Introduction
Complex diseases are a devastating consequence of usually unknown environmental factors and the combined effects of tens to thousands of genetic variants that are spread throughout the genome.1 The advanced use of bioinformatic tools will provide a better understanding of the intricate network of multiple genetic effects that shapes the architecture of complex diseases.2
Immune-mediated inflammatory diseases (IMIDs) comprise a variety of complex diseases characterised by the loss of self-tolerance, the maintenance of chronic inflammation and an aberrant immune response.3 Genome-wide association studies (GWAS) have largely increased our understanding of the aetiology of complex diseases, providing new data about the genome and lighting the way to the identification of genes and pathways that contribute to disease susceptibility and prognosis. Many susceptibility loci have been discovered for IMIDs, and several are shared between diseases, adding a common genetic background to their overlapping clinical and immunological characteristics.4 Additionally, GWAS findings have also confirmed that the contribution of each associated locus to disease risk is often small and has low predictive value.1
To address complex disease susceptibility, three main components must be considered: genetics, environmental exposures and lifestyle factors.1 4 As for genetics, large cohorts have been genotyped in GWAS efforts, and hundreds of genetic risk factors have been identified.5 However, GWAS data can be examined in various ways, moving forward to a more precise genetic profiling, its use for personalised medicine and the identification of individuals with higher risk of displaying a specific phenotype.6 Genomic Risk Scores (GRS) take into account disease heritability and the additive effect of genetic polymorphisms, and they provide a disease risk score per individual to evaluate their relative risk to suffer a disease.7–9
GRS are calculated essentially by combining the weighted effects of the risk alleles for each individual; these weighted effects are assigned depending on the strength of the association to the risk of disease—the effect size.7 10 The identification of individuals with high risk or those prone to developing more aggressive phenotypes is a useful tool for personalised medicine and clinical management of patients. GRS have been successful in several diseases such as schizophrenia11 and obesity.12 This strategy had a great impact on cardiovascular diseases such as coronary artery disease12–14 but also in IMIDs such as sarcoidosis,15 systemic lupus erythematosus (SLE)16 17 and vitiligo18 recently.
Systemic sclerosis (SSc) or scleroderma is a complex chronic autoimmune disease. It belongs to the group of IMIDs and it has one of the highest mortality rates among them.19 SSc affects the connective tissue and shows complex and varied clinical manifestations. Raynaud’s phenomenon and gastro-oesophageal reflux are two common onset symptoms, but they are not exclusive to SSc. Conversely, the disease can manifest in different ways, such as affectation of the skin (inflammatory skin disease, extensive fibrosis), musculoskeletal inflammation and vascular damage.20–22 Furthermore, SSc also shows organ-specific manifestations, such as lung fibrosis, pulmonary arterial hypertension, renal failure and gastrointestinal complications. Notably, the involvement of the lungs, with pulmonary hypertension and/or pulmonary fibrosis, is the leading cause of death in SSc.19
Patients with SSc can be classified into different subgroups according to clinical outcome: limited cutaneous scleroderma (lcSSc) or diffuse cutaneous scleroderma (dcSSc), depending on how widespread fibrosis is.23 On other hand, they can also be classified depending on their serological status, considering the presence of the mutually exclusive anti-centromere or anti-topoisomerase autoantibodies (ie, ACA+ or ATA+).22 23
Since the first SSc GWAS in European populations was carried out 10 years ago,24 our recently published meta-GWAS is the largest effort to decipher the genetic component of SSc.25 In addition to the extensively known association of the human leucocyte antigen (HLA) region with the disease, 27 non-HLA GWAS level associations and 3 suggestive loci were identified.25
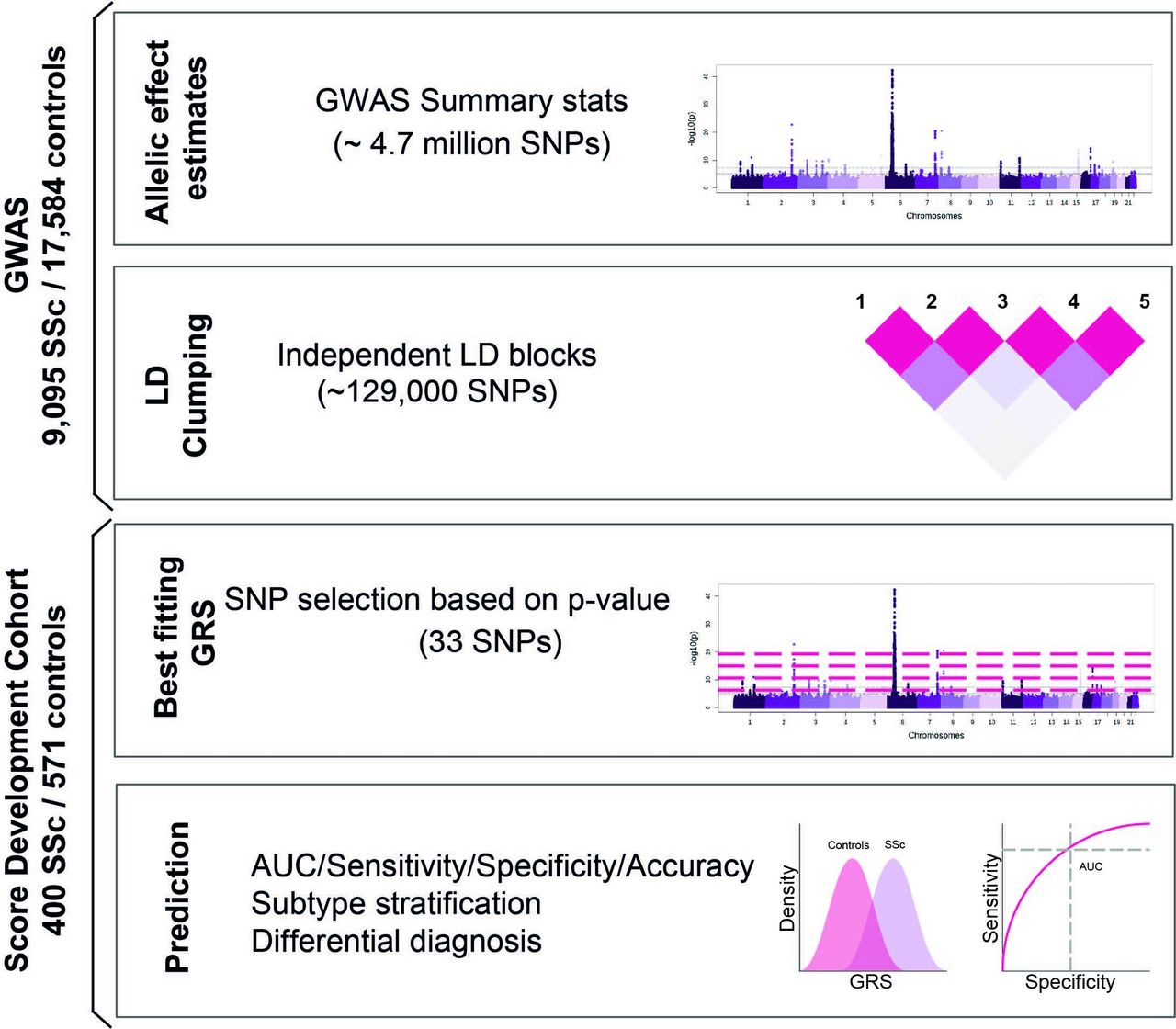
Considering the heterogeneity and variable prognosis of patients with SSc, GRS could be a powerful tool in clinical diagnosis to identify patients in the early stages of the disease and to differentiate them from patients with confounding diseases. By taking advantage of the summary statistics of this large meta-GWAS, we generated an accurate SSc GRS through the use of an independent and unique dataset comprising patients with SSc and with other IMIDs3 (figure 1). We generated subtype-specific GRS for the clinical and serological SSc subgroups of patients, and we tested the clinical implications of GRS in SSc. Finally, the GRS was complemented with additional demographic and immunological information.

Overview of the study design. AUC, area under the receiver operating characteristic (ROC) curve; GRS, Genomic Risk Scores; GWAS, genome-wide association studies;SNPs, single nucleotide polymorphisms.
Methods
GRS calculation
GRS was developed as implemented in PRSice-2,26 using summary statistics and assuming an additive effect for the effective allele. Briefly, PRSice-2 calculated the product of the number of effect alleles per individual and the respective SNP weights. The score was averaged by the number of alleles included in the GRS per individual (argument --score avg). We used the minor allele frequency in the PRECISESADS cohort as the genotype for the samples with missing genotype. We applied a 10 000 permutation procedure to calculate the empirical p value (--perm 10 000).
PRSice-2 allowed us to fit different GRS models by selecting only the variants that passed a number of different p value thresholds in the GWAS summary statistics (argument --bar-levels 5e-11, 5e-10, 5e-09, 5e-08, 5e-07, 5e-06, 5e-05, 0.0001, 0.001, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 1, but GRS calculated at all intermediate p value thresholds, high resolution parameters, were calculated) using sex (female/male) as covariate. Therefore, the model fit is defined as: R2 of the full model (SSc case or control ~ GRS + Sex) − R2 of the null model (SSc case or control ~ Sex).
Multivariate model
In order to test if a combination of GRS with demographic factors and the counts of immune cell subpopulations in peripheral blood would improve the predictive value of our model, we divided our score development cohort into an initial set, comprising the non-Spanish individuals in the PRECISESADS study (n=518), in which we developed a multivariate model and a testing set that comprised all the Spanish individuals in this study (n=339).
First, we built several generalised linear models that included GRS and each demographic and immune parameter in online supplemental table 1 individually, then we compared them to the null model that included only GRS and sex as covariates. Improvement over the null model was defined by an LRT (p value<0.05).
Supplemental material
Second, we generated a multivariate model that included the 13 phenotypic variables that had been identified as informative in the previous step. Using leave-one-out prediction (ie, including all variables but one in the model) and comparing to the full model, we calculated the contribution of all variables to the multivariate model. This model was applied to the testing set of individuals.
Details about the cohorts, linkage disequilibrium (LD) clumping, GRS additive model, the model fitting analyses and the effects of including country of origin as covariates are shown in the online supplemental methods section.
Supplemental material
Results
A 33-variant GRS discriminates between patients with SSc and controls
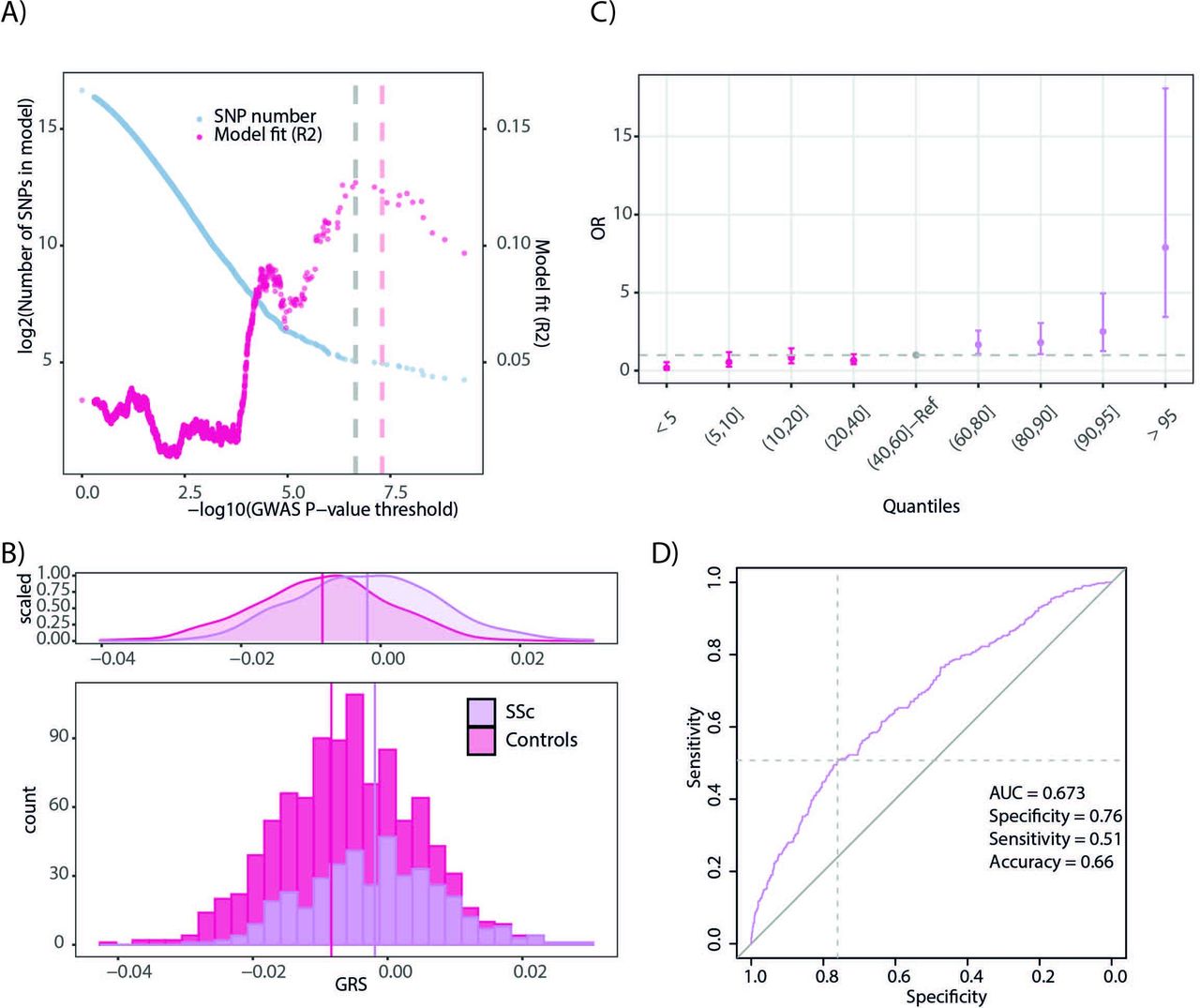
We calculated GRS in an independent score development cohort comprising 400 patients with SSc and 571 healthy controls.27 We observed that the best-fitting GRS (GRS R2=0.13; p value=1.27×10-17; permutation p value=9.99×10-5) included 33 independent SNPs that had a p value<2.215×10-7 (figure 2A). Sex, which was included as a covariate, contributed very modestly to the explained variance (R2=0.01).

Systemic sclerosis Genomic Risk Scores (SSc GRS). (A) Identification of the best-fitting GRS in the score development cohort. Tested p value thresholds for the SNPs included in the GWAS summary statistics are presented in the x-axis. The number of SNPs included in the models corresponding to each p value threshold is shown on the left y-axis. Model fit (R2) is represented in the right y-axis. (B) Distribution of GRS for patients with SSc and healthy controls in the score development cohort. (C) Relative risk for individuals in different quantiles of the GRS distribution. (D) receiver operating characteristic (ROC) curve for the 33 SNP SSc GRS. AUC, area under the ROC curve; GWAS, genome-wideassociation studies; SNPs, single nucleotide polymorphisms.
As expected, the SSc cases and controls showed significantly different GRS distributions (figure 2B, control group mean=−8.35×10-3 and SSc group mean=−1.91×10-3, t-test p value<2.2×10-16). Reassuringly, individuals with GRS in the 95th percentile showed a fivefold higher relative risk (OR=7.89, 95% CI 3.44 to 18.08) than the reference quantile (40th–60th percentiles) (figure 2C).
Reassuringly, the 33 variant GRS had a 67% chance of accurately predicting if an individual was a patient with SSc or an unaffected control (AUC=0.673, 95% CI 0.64 to 0.71, p value=3.90×10-23, figure 2D). We determined a best-fitting GRS threshold (GRS controls<−1.86×10-3<GRS cases, details in online supplemental methods) and reached a moderate discrimination between cases and controls (specificity=0.76; sensitivity=0.51; accuracy=0.66, figure 2D).
We observed that if the receiver operating characteristic (ROC) curves were calculated separately for each country of origin, the AUC determined by the 33 variant GRS ranged from 0.60 to 0.75 (online supplemental figure 2A). However, variability of the AUC did not correlate with either country longitude, latitude or distance to 1000 genomes GBR and CEU populations (see online supplemental methods, online supplemental figure 2B-D).
Supplemental material
Subtype stratified SSc GWAS summary stats discriminate between clinical and serological subtypes
The 33 variant GRS previously described distinguished between patients with SSc and healthy controls. However, SSc is a heterogeneous disease with both clinical and serological subtypes that influence the prognosis of the disease, and the prediction of these subtypes is a major clinical demand. The 33 SNP SSc GRS showed no predictive value for clinical subtypes (dcSSc vs lcSSc AUC=0.496, 95% CI 0.40 to 0.59, p value=0.93, online supplemental figure 3) and serological subtypes (ATA+ vs ACA+ AUC = 0.464, 95% CI 0.37 to 0.56, p value=0.45, online supplemental figure 3). Furthermore, this SSc GRS was not able to predict the development of pulmonary fibrosis in patients with SSc (SSc with pulmonary fibrosis vs SSc without pulmonary fibrosis AUC=0.479, 95% CI 0.38 to 0.57, p value=0.66, online supplemental figure 3).
Supplemental material
Therefore, we used the allelic effects obtained in the GWAS comparison between dcSSc and lcSSc and between ATA+ and ACA+ patients to build subtype-specific GRS. The best-fitting GRS p value threshold for the variants in the dcSSc versus lcSSc comparison, clinical subtype GRS, comprised up to 9780 SNPs (SNP p value threshold for the best- fitting dcSSc vs lcSSc GRS <9.99×10-2, figure 3A). This clinical subtype GRS was not limited to highly significant variants but it also included thousands of additional SNPs with very low significance. The GRS for the variants in the ATA+ vs ACA+ comparison, serological subtype GRS, required up to 35 058 SNPs (SNP p value threshold for the best-fitting ATA+ vs ACA+ GRS < 3.48×10-1, figure 3A). The clinical subtype GRS did not explain much of the phenotypic variance between dcSSc and lcSSc (R2=0.053), while the explained variance between them using the serological subtype GRS was comparable with the SSc GRS (R2=0.115). In this context, the subtype-specific GRS distributions (mean dcSSc GRS=2.46×10-3; mean lcSSc GRS=2.16×10-3; t-test p value=1.21×10-2, figure 3B), and AUC based on the clinical subtype GRS led to a modest classification of the patients into the dcSSc or lcSSc groups (AUC=0.604, 95% CI 0.51 to 0.70, p value=2.59×10-2, figure 3C). However, the serological subtype GRS (comprising 35 058 SNPs) showed more distinctive GRS distributions between ATA+ and ACA+ patients (mean ATA+ GRS = 1.39×10-3 and mean ACA+ GRS=1.11×10-3, t-test p value=1.12×10-4, figure 3B), and best classification results for the ATA+ or ACA+ subgroups of patients (AUC=0.693, 95% CI 0.61 to 0.78, p value=7.58×10-6, figure 3C).

Characteristics of clinical subtype-specific Genomic Risk Scores (GRS) (left) and serological subtype-specific GRS (right). (A) Identification of the best-fitting GRS in the score development cohort. Tested p value thresholds for the SNPs included in the GWAS summary statistics are presented in the x-axis. The number of SNPs included in the models corresponding to each p value threshold is shown on the left y-axis. Model fit (R2) is represented in the right y-axis. (B) Distribution of GRS for patients with systemic sclerosis (SSc) in each subtype group. (C) Receiver operating characteristic (ROC) curves for the 9 780 SNP clinical subtype-specific GRS and 35 058 SNP serological subtype-specific GRS. AUC, area under the ROC curve; SNPs, single nucleotide polymorphisms.
Considering the clinical relevance of pulmonary fibrosis for the prognosis of patients with SSc, we tested the predictive value of both the clinical and the serological GRS on the development of lung fibrosis. Interestingly, we observed that the serological GRS was marginally able to discriminate between patients with and without lung fibrosis but the model did not reach statistical significance (AUC=0.575, 95% CI 0.48 to 0.67, p value=0.11, online supplemental figure 3).
GRS separates SSc from other IMIDs
Considering the shared genetic component of IMIDs, the implementation of the proposed GRS might help to identify high-risk individuals not only for SSc but also for other immune-related traits. Regarding the accuracy of the 33 variant SSc GRS in other IMIDs, we observed that the SSc GRS was able to separate patients with RA (RA group mean=−4.46×10-3; t-test p value<2.8×10-9), Sjögren syndrome, SJS (SJS group mean=−1.78×10-3; t-test p value<3.54×10-6) and SLE (SLE group mean=−3.67×10-3; t-test p value<8.51×10-13) from the non-affected individuals. However, as expected, the GRS differences between patients with RA, SJS and SLE and controls were less significant than between SSc cases and controls (figure 4A). Furthermore, using the SSc GRS in these three additional IMIDs, the AUCs showed a modest predictive value (AUC RA=0.608, 95% CI 0.57 to 0.64, p value=6.58×10-9; SJS=0.590, 95% CI 0.55 to 0.63, p value=1.58×10-6; AUC SLE=0.623, 95% CI 0.59 to 0.66, p value=3.94×10-12, figure 4B).

Impact of the 33 SNP systemic sclerosis (SSc) Genomic Risk Scores (GRS) on the differential classification with other immune-mediated inflammatory diseases IMIDs). (A) Distribution of GRS for healthy controls and patients with SSc, systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and Sjögren syndrome (SJS). (B) Receiver operating characteristic (ROC) curves for the predictive value of the SSc GRS to distinguish between patients with SSc, SLE, RA or SJS and healthy controls. (C) ROC curves for the predictive value of the SSc GRS to distinguish between patients with SLE, RA or SJS and patients with SSc. AUC, area under the ROC curves.
A key point toward GRS being implemented from bench-to-bedside is not only the ability to identify individuals at high risk of developing SSc in the general population, but also to help in the differential diagnosis between SSc and other IMIDs. In the pursuit of this objective, we tested the effectiveness of our SSc GRS to correctly classify between patients with SSc and those affected by other IMIDs. We report statistical differences between the GRS distributions for SSc and rheumatoid arthritis (RA) (t-test p value<3.78×10-4) or SJS (t-test p value<3.70×10-6), but only nominally significant differences in the case of SLE (t-test p value<1.37×10-2) (figure 4A). These results were aligned with the predictive capacity of the GRS in the separation between patients with SSc and other IMIDs. The greatest AUC was observed for the classification of patients with SSc versus patients with SJS (SJS AUC=0.585, 95% CI 0.55 to 0.62, p value=2.22×10-5), and decreased in more closely related IMIDs, such as RA (AUC RA=0.568, 95% CI 0.53 to 0.61, p value=8.84×10-4) and, especially, SLE (SLE AUC=0.553, 95% CI 0.51 to 0.59, p value=1.19×10-2) (figure 4C).
Age and immune cell counts improve the prediction accuracy
The score development cohort recruited in the PRECISESADS study was comprehensively phenotyped and allowed us to complement our GRS with additional demographic (age, sex) and immunological (immune cell counts in peripheral blood estimated using a large flow cytometry panel) parameters28 (online supplemental table 1). We divided our score development cohort into an initial set (n=518) and a testing subgroup (n=339). The initial set allowed us to test the relevance of the different parameters in a combined GRS and phenotypic model. On the other hand, the testing set confirmed these findings.
First, we identified the demographic and immunological parameters which improved the GRS model (LRT p value<0.05) (online supplemental table 1). Twelve immune cell subtypes in peripheral blood showed a significant contribution to the model, but the most significant contribution among the phenotypic variables corresponded to age (LRT p value=3.47×10-20, online supplemental table 2).
When we combined only the informative variables into the same model, multivariate GLM, in addition to GRS and age, only 4 out of the 12 immune cell types remained as independently associated in the multivariate model: resting NK cells, M0 macrophages, activated dendritic cells and memory B cells (online supplemental table 3). The contribution of sex to the model did not remain significant when considering all the independent variables together and GRS score distributions between male and female patients did not show significant information (t-test p value=0.24, online supplemental table 3). Using leave-one-out prediction, we identified age as the most informative variable, followed by GRS (online supplemental table 4). We observed that the contribution of GRS to the model was comparable with the contribution of all significant parameters of immune cell count together (GRS LRT p value=2.59×10-12; GRS LRT p value=1.26×10-12, online supplemental table 4).
The multivariate GLM described above (SSc status ~GRS+Age+Memory B cells+Resting NK cells+M0 Macrophages+Activated dendritic cells) greatly outperformed the GRS and sex only model both in the initial (AUC discovery=0.847, 95% CI 0.81 to 0.88, p value=1.10×10-90) and in the testing set (AUC=0.787, 95% CI 0.73 to 0.84, p value=1.31×10-24), as illustrated in figure 5. Moreover, the multivariate GLM outperformed the models that did not include age, GRS or both (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Receiver operating characteristic (ROC) curves for the predictive value of the multivariate generalised linear model (GLM), (SSc status ~GRS+Age+Memory B cells+Resting NK cells+M0 Macrophages+Activated dendritic cells) to distinguish between patients with SSc and healthy controls in the initial and replication cohorts depending on the variables included in the models. GRS, Genomic Risk Scores; NK cells, natural killer cells; SSc, systemic sclerosis.
Discussion
We generated a GRS based on the allelic effects identified in the largest GWAS in SSc to date.25 We obtained a predictive GRS model comprising 33 genetic polymorphisms, which allowed us to differentiate between SSc and controls in an independent SSc patient cohort.27 A serological subtype-specific GRS (based on the GWAS comparison between ATA+ and ACA+ SSc patients) showed the best predictive value to classify patients based on the presence of different autoantibodies. Furthermore, we demonstrated the accuracy of the model in the differentiation between SSc and other IMIDs, such as RA and SJS. Finally, we complemented the SSc GRS with demographic data and peripheral blood immune cell counts in a multivariate model which reached a very significant recall rate.
The reported SSc GRS showed good predictive value (AUC=0.673), in line with the GRS developed for other IMIDs. For example, a similar AUC was reported for inflammatory bowel disease with a GRS based on the allelic effects observed for 12 882 cases and 132 532 healthy controls (AUC=0.7229) and in SLE (AUCs ranging 0.62–0.78.16 17 Moreover, Stahl et al implemented a Bayesian inference model in a GWAS that comprised 5 485 cases of RA and 22 609 healthy controls, and the model explained 18% of the total variance, which is comparable to the variance explained by our model (R2=0.13).30 We would like to note that the previously conducted GWAS comprised 9 095 SSc cases and 17 584 controls, and the SSc GRS was developed in an independent cohort of 400 patients with SSc and 571 non-affected controls recruited for the PRECISESADS project.27 Since sample size is key in the identification of reliable genetic association signals and in the accurate estimation of allelic effects in GWAS,6 7 31 the presented SSc GRS represents a robust model supported by substantial statistical power. Nevertheless, despite the promising results of the described SSc GRS, the sensitivity and specificity of the model are still far from clinical use and it will require the addition of extra information and/or the development of well-powered phenotype-specific GWAS to identify cases with specific phenotypes with higher statistical power.
Furthermore, we consider that the SSc GRS is not heavily influenced by LD clumping, since we included only the top HLA SNP association in the GRS in order to avoid an over-representation of HLA polymorphisms without discarding completely the potential of this region in GRS. Nevertheless, it should be noted that all the samples included in the GWAS summary stats and in the score development cohorts for the SSc GRS had European ancestry25 27 (online supplemental figure 1). One of the major limitations of GRS implementation is the bias toward populations with a similar ethnic origin to the discovery sample, that is, the GRS shows better accuracy in closely related populations.7 32 As we illustrated in online supplemental figure 2, we found differences in the AUCs reached by the SSc GRS in the score development cohort depending on the origin of the individuals. Consequently, the performance of the SSc GRS in non-European or mixed populations should be taken with caution.7 33
Supplemental material
A possible confounding factor for GRS in IMIDs is the shared genetic and immunological component that makes diagnosis complex and a slow clinical process especially in the early stages of these diseases.34–36 As a clinical tool, a robust GRS improves early diagnosis and helps in differential diagnosis.31 Although the accuracy of the SSc GRS in differentiating between SSc and other IMIDs is still far from clinical standards, the model was able to discriminate between SSc and RA in 56.8% of the cases, and between SSc and SJS in 58.5% of the cases (figure 4). However, for SLE and SSc, which have a well-documented shared genetic component,3 35 it was not possible to reach an accuracy that allowed for case differentiation. Taking into account the above, we consider that the reported GRS could enhance SSc diagnosis in the future and may contribute to personalised medicine, as a tool to assist physicians in the diagnosis of SSc.
In addition to comorbidities with other IMIDs, there is great variability in the disease course followed by patients with SSc, since their treatment and prognosis in the long term is very heterogeneous.20 Chen et al 17 developed a GRS based on a GWAS analysing patients with SLE with and without renal involvement, but this SLE nephritis-specific GRS did not outperform the SLE severity predictions achieved with a SLE GRS. Following a similar strategy, we generated two additional GRS based on the GWAS comparisons between clinical and serological subtypes in patients with SSc. Remarkably, we showed that the serological subtype-specific GRS was able to differentiate SSc cases within the serological subtypes (ACA+ or ATA+), which is a promising result in the use of GRS to predict prognosis in SSc.37 Regarding specific clinical outcomes, we focused on the use of GRS to predict lung fibrosis due to the disastrous effect of lung involvement on the survival of patients with SSc. We could not use SSc lung involvement GWAS data, but we observed that the serological subtype-specific GRS allowed us to correctly infer the existence of lung fibrosis on patients with SSc in 57.5% of cases (online supplemental figure 3).
Finally, we explored the possibilities of combining GRS with demographic and immunological covariates. We found that, out of all the covariates tested, age and the relative abundance of different immune cell types proved to be informative and resulted in a higher sensitivity in the case/control classification. As expected, age was confirmed as a very relevant factor in our model. Age is known to influence SSc, since patients with SSc are often diagnosed in their midlife ages.19 38 On the other hand, sex was included as a covariate to calculate the best p value threshold for the GRS and in the multivariate model, but, in both cases, it was not very informative. This lack of significant contribution of sex to the GRS model was also reported previously in SLE.17 Therefore, these counterintuitive results for a known SSc risk factor19 were likely due to the selection of a sex-matched control population (online supplemental table 1), which would rule out the relevance of this parameter. The immune cell types included in the multivariate GRS were also concordant with the known aetiopathogenesis of the disease.22 Functional defects or genetic susceptibility variants located in relevant genes for dendritic cells, macrophages and B cells have been described in patients with SSc.39–43 T cell subtypes were relevant covariates in the model initially, but no T cell subset was selected for the multivariate model (online supplemental tables 2–4). Considering the central role of T cells in SSc, we hypothesise that since we could not include the Th1, Th2 or Th17 fractions in the model, this effect might have been overlooked.43
We have generated a GRS using a GWAS dataset and a score development cohort in which training was carried out and empirical p values for the GRS were obtained via permutation. Therefore, although both cohorts were independent, out-of-sample prediction has not been performed and it is a limitation of the present study. Consequently, our model and results should be considered as seminal work for future validation in additional cohorts of patients with SSc.
In summary, we developed a GRS based on the largest GWAS in SSc, resulting in a sensitive model to differentiate between SSc cases and non-affected controls, but also to differentiate within the different SSc serological subtypes (ATA+ and ACA+). Additionally, the GRS was also useful to differentiate patients with SSc from those affected by RA and SJS. We have shown that the GRS strategy in SSc has great potential to contribute to the field. However, several limitations and challenges, such as non-European ancestry or sample size, must be overcome to implement this strategy in clinical management.
Acknowledgments
This work is part of the PhD thesis entitled Deciphering the genetic basis of systemic sclerosis by GV-M. We would like to thank Sofia Vargas and Gemma Robledo for their excellent technical assistance. We also appreciate the controls and the affected individuals who generously provided the samples for these studies.
Footnotes
LB-C and GV-M are joint first authors.
Handling editor Josef S Smolen
Twitter @ShervinAssassi
LB-C and GV-M contributed equally.
Collaborators International SSc Group: P. Carreira, Department of Rheumatology, 12 de Octubre University Hospital, Madrid, Spain; I. Castellvi, Department of Rheumatology, Santa Creu i Sant Pau University Hospital, Barcelona, Spain; R. Ríos, Department of Internal Medicine, San Cecilio Clinic University Hospital, Granada, Spain; J. L. Callejas, Department of Internal Medicine, San Cecilio Clinic University Hospital, Granada, Spain; R. García Portales, Department of Rheumatology, Virgen de la Victoria Hospital, Málaga, Spain; A. Fernández-Nebro, Department of Rheumatology, Carlos Haya Hospital, Málaga, Spain; F. J. García-Hernández, Department of Internal Medicine, Virgen del Rocío Hospital, Sevilla, Spain; M. A. Aguirre, Department of Rheumatology, Reina Sofía/IMIBIC Hospital, Córdoba, Spain; B. Fernández-Gutiérrez, Department of Rheumatology, San Carlos Clinic Hospital, Madrid, Spain; L. Rodríguez-Rodríguez, Department of Rheumatology, San Carlos
Clinic Hospital, Madrid, Spain; P. García de la Peña, Department of Rheumatology, Madrid Norte Sanchinarro Hospital, Madrid, Spain; E. Vicente, Department of Rheumatology, La Princesa Hospital, Madrid, Spain; J. L. Andreu, Department of Rheumatology, Puerta de Hierro Hospital-Majadahonda, Madrid, Spain; M. Fernández de Castro, Department of Rheumatology, Puerta de Hierro Hospital-Majadahonda, Madrid, Spain; F. J. López-Longo, Department of Rheumatology, Gregorio Marañón University Hospital, Madrid, Spain; V. Fonollosa, Department of Internal Medicine, Valle de Hebrón Hospital, Barcelona, Spain; A. Guillén, Department of Internal Medicine, Valle de Hebrón Hospital, Barcelona, Spain; G. Espinosa, Department of Internal Medicine, Clinic Hospital, Barcelona, Spain; C. Tolosa, Department of Internal Medicine, Parc Tauli Hospital, Sabadell, Spain; A. Pros, Department of Rheumatology, Hospital Del Mar, Barcelona, Spain; M. Rodríguez Carballeira, Department of Internal Medicine, Hospital Universitari Mútua Terrasa, Barcelona, Spain; F. J. Narváez, Department of Rheumatology, Bellvitge University Hospital, Barcelona, Spain; M. Rubio Riva, Department of Internal Medicine, Bellvitge University Hospital, Barcelona, Spain; V. Ortiz-Santamaría, Department of Rheumatology, Granollers Hospital, Granollers, Spain; A. B. Madroñero, Department of Internal Medicine, Hospital General San Jorge, Huesca, Spain; M. A. González-Gay, Epidemiology, Genetics and Atherosclerosis Research Group on Systemic Inflammatory Diseases, DIVAL, University of Cantabria, Santander, Spain; B. Díaz, Department of Internal Medicine, Hospital Central de Asturias, Oviedo, Spain; L. Trapiella, Department of Internal Medicine, Hospital Central de Asturias, Oviedo, Spain; M. V. Egurbide, Department of Internal Medicine, Hospital Universitario Cruces, Barakaldo, Spain; P. Fanlo-Mateo, Department of Internal Medicine, Hospital Virgen del Camino, Pamplona, Spain; L. Saez-Comet, Department of Internal Medicine, Hospital Universitario Miguel Servet, Zaragoza, Spain; F. Díaz, Department of Rheumatology, Hospital Universitario de Canarias, Tenerife, Spain; E. Beltrán, Department of Rheumatology, Hospital General Universitario de Valencia, Valencia, Spain; J. A. Roman-Ivorra, Department of Rheumatology, Hospital Universitari i Politecnic La Fe, Valencia, Spain; J. J. Alegre Sancho, Department of Rheumatology, Hospital Universitari Doctor Peset, Valencia, Spain; M. Freire, Department of Internal Medicine, Thrombosis and Vasculitis Unit, Complexo Hospitalario Universitario de Vigo, Vigo, Spain; F. J. Blanco Garcia, Department of Rheumatology, INIBIC-Hospital Universitario A Coruña, La Coruña, Spain; N. Oreiro, Department of Rheumatology, INIBIC-Hospital Universitario A Coruña, La Coruña, Spain; T. Witte, Department of Clinical Immunology, Hannover Medical School, Hannover, Germany; A. Kreuter, Department of Dermatology, Josefs-Hospital, Ruhr University Bochum, Bochum, Germany; G. Riemekasten, Clinic of Rheumatology, University of Lübeck, Lübeck, Germany; P. Airo, Service of Rheumatology and Clinic Immunology Spedali Civili, Brescia, Italy; C. Magro, Department of Rheumatology, Leiden University Medical Center, Leiden, The Netherlands; A. E. Voskuyl, Department of Rheumatology, VU University Medical Center, Amsterdam, The Netherlands; M. C. Vonk, Department of Rheumatology, Radboud University Nijmegen Medical Center, Nijmegen, Netherlands; R. Hesselstrand, Department of Rheumatology, Lund University, Lund, Sweden; A. Nordin, Division of Rheumatology, Department of Medicine, Karolinska University Hospital, Karolinska Institute, Stockholm, Sweden; A. L. Herrick, Centre for Musculoskeletal Research, The University of Manchester, Salford Royal NHS Foundation Trust, Manchester Academic Health Science Centre, Manchester, UK, NIHR Manchester Biomedical Research Centre, Manchester, UK; C. Lunardi, Department of Medicine, Università degli Studi di Verona, Verona, Italy; G. Moroncini, Clinica Medica, Department of Clinical and Molecular Science, Università Politecnica delle Marche and Ospedali Riuniti, Ancona, Italy; A. Hoffmann-Vold, Department of Rheumatology, Oslo University Hospital, Oslo, Norway; J. H. W. Distler, Department of Internal Medicine 3, Institute for Clinical Immunology, University of Erlangen-Nuremberg, Erlangen, Germany; L. Padyukov, Division of Rheumatology, Department of Medicine, Karolinska University Hospital, Karolinska Institute, Stockholm, Sweden; B. P. C. Koeleman, University Medical Center Utrecht, Utrecht, The Netherlands. Australian Scleroderma Interest Group (ASIG): J. Zochling, Menzies Research Institute Tasmania, University of Tasmania, Hobart, TAS, Australia; J. Sahhar, Department Rheumatology, Monash Medical Centre, Melbourne, VIC, Australia; J. Roddy, Rheumatology, Royal Perth Hospital, Perth, WA, Australia; P. Nash, Research Unit, Sunshine Coast Rheumatology, Maroochydore, QLD, Australia; K. Tymms, Canberra Rheumatology, Canberra, ACT, Australia; M. Rischmueller, Department Rheumatology, The Queen Elizabeth Hospital,Woodville, SA, Australia; S. Lester, Department Rheumatology, The Queen Elizabeth Hospital,
Woodville, SA, Australia; S. Proudman, Royal Adelaide Hospital and University of Adelaide, Adelaide, SA, Australia; W. Stevens, St. Vincent’s Hospital, Melbourne, VIC, Australia; M. Nikpour, The University of Melbourne at St. Vincent’s Hospital, Melbourne, VIC, Australia; M. A. Brown, Institute of Health and Biomedical Innovation, Queensland University of Technology,
Translational Research Institute, Princess Alexandra Hospital, Brisbane, QLD, Australia. PRECISESADS Clinical Consortium: Doreen Belz, Klinik und Poliklinik für Dermatologie und Venerologie, Uniklinik Köln, Köln, Germany; Francesca Ingegnoli, Department of Clinical Sciences and Community Health, University of Milan, Milan, Italy; Yolanda Jimenez Gómez, IMIBIC, Reina Sofia Hospital, University of Cordoba, Cordoba, Spain; Chary Lopez Pedrera, IMIBIC, Reina Sofia Hospital, University of Cordoba, Cordoba, Spain; Rik Lories, Division of Rheumatology, University Hospitals Leuven and Skeletal Biology and Engineering Research Center, KU Leuven, Leuven, Belgium; Eduardo Collantes-Estevez, IMIBIC, Reina Sofia Hospital, University of Cordoba, Cordoba, Spain; Gaia Montanelli, Scleroderma Unit, Referral Center for Systemic Autoimmune Diseases, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico di Milano, Milan, Italy; Silvia Piantoni, Immunology & Allergy, University Hospital and School of Medicine, Geneva, Switzerland; Ignasi Rodriguez Pinto, Division of Rheumatology, University Hospitals Leuven and Skeletal Biology and Engineering Research Center, KU Leuven, Leuven, Belgium; Carlos Vasconcelos, Serviço de Imunologia EX-CICAP, Centro Hospitalar e Universitário do Porto, Porto, Portugal. PRECISESADS Flow Cytometry study group: Christophe Jamin, 1U1238, Université de Brest, Inserm, Labex IGO, CHU de Brest, Brest, France; Concepción Marañón, GENYO, Centre for Genomics and Oncological Research Pfizer, University of Granada, Andalusian Regional Government, PTS GRANADA, Granada, Spain; Lucas Le Lann, 1U1238, Université de Brest, Inserm, Labex IGO, CHU de Brest, Brest, France; Quentin Simon, 1U1238, Université de Brest, Inserm, Labex IGO, CHU de Brest, Brest, France; Bénédicte Rouvière, 1U1238, Université de Brest, Inserm, Labex IGO, CHU de Brest, Brest, France; Nieves Varela, GENYO, Centre for Genomics and Oncological Research Pfizer, University of Granada, Andalusian Regional Government, PTS GRANADA, Granada, Spain; Brian Muchmore, GENYO, Centre for Genomics and Oncological Research Pfizer, University of Granada, Andalusian Regional Government, PTS GRANADA, Granada, Spain; Aleksandra Dufour, Immunology & Allergy, University Hospital and School of Medicine, Geneva, Switzerland; Montserrat Alvarez, Immunology & Allergy, University Hospital and School of Medicine, Geneva, Switzerland; Jonathan Cremer, Laboratory of Clinical Immunology, Department of Microbiology and Immunology, KU Leuven, Leuven, Belgium; Nuria Barbarroja, IMIBIC, Reina Sofia Hospital, University of Cordoba, Cordoba, Spain; Velia Gerl, Department of Rheumatology and Clinical Immunology, Charité University Hospital, Berlin, Germany; Laleh Khodadadi, Department of Rheumatology and Clinical Immunology, Charité University Hospital, Berlin, Germany; Qingyu Cheng, Department of Rheumatology and Clinical Immunology, Charité University Hospital, Berlin, Germany; Anne Buttgereit, Bayer AG, Berlin, Germany; Aurélie De Groof, Pôle de Pathologies Rhumatismales Inflammatoires et Systémiques, Institut de Recherche Expérimentale et Clinique, Université catholique de Louvain, Brussels, Belgium; Julie Ducreux, Pôle de Pathologies Rhumatismales Inflammatoires et Systémiques, Institut de Recherche Expérimentale et Clinique, Université catholique de Louvain, Brussels, Belgium; Elena Trombetta, Laboratorio di Analisi Chimico Cliniche e Microbiologia - Servizio di Citofluorimetria, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico di Milano, Milano, Italy; Tianlu Li, Chromatin and Disease Group, Bellvitge Biomedical Research Institute (IDIBELL), Barcelona, Spain; Damiana Alvarez-Errico, Chromatin and Disease Group, Bellvitge Biomedical Research Institute (IDIBELL), Barcelona, Spain; Torsten Witte, Klinik für Immunologie und Rheumatologie, Medical University Hannover, Hannover, Germany; Katja Kniesch, Klinik für Immunologie und Rheumatologie, Medical University Hannover, Hannover, Germany; Nancy Azevedo, GENYO, Centre for Genomics and Oncological Research Pfizer, University of Granada, Andalusian Regional Government, PTS GRANADA, Granada, Spain; Esmeralda Neves, GENYO, Centre for Genomics and Oncological Research Pfizer, University of Granada, Andalusian Regional Government, PTS GRANADA, Granada, Spain; Nancy Azevedo, IMIBIC, Reina Sofia Hospital, University of Cordoba, Cordoba, Spain and Serviço de Imunologia EX-CICAP, Centro Hospitalar e Universitário do Porto, Porto, Portugal; Esmeralda Neves, IMIBIC, Reina Sofia Hospital, University of Cordoba, Cordoba, Spain and Serviço de Imunologia EX-CICAP, Centro Hospitalar e Universitário do Porto, Porto, Portugal; Sambasiva Rao, Sanofi Genzyme, Framingham, MA, USA; Pierre-Emmanuel Jouve, AltraBio SAS, Lyon, France.
Contributors LB-C: data analysis, manuscript drafting, revision and approval; GV-M: data analysis, manuscript drafting, revision and approval; MK: interpretation of data, manuscript revision and approval; MA-H: interpretation of data, manuscript revision and approval; ELI: data interpretation, manuscript revision and approval; PRECISESADS Clinical Consortium: data acquisition, manuscript revision and approval; MEAl-R: data interpretation, manuscript revision and approval; LB: data interpretation, manuscript revision and approval; JM: study design, manuscript drafting, revision and approval.
Funding This work was supported by the Spanish Ministry of Science and Innovation (grant ref. RTI2018101332-B-100), Red de Investigación en Inflamación y Enfermedades Reumáticas (RIER) from Instituto de Salud Carlos III (RD16/0012/0013), and EU/EFPIA/Innovative Medicines Initiative Joint Undertaking PRECISESADS grant no. 115565. LBC and MAH were funded by the Spanish Ministry of Science and Innovation through the Juan de la Cierva incorporation program (ref. IJC2018-038026-I and IJC2018-035131-I, respectively). GV-M was funded by the Spanish Ministry of Science and Innovation through the Ayudas para contratos predoctorales para la formación de doctores 2019 program (ref. RTI2018-101332-B-I00).
Competing interests LB-C: none; GV-M: none; MK: none; MA-H: none; ELI: none; International SSc Group: none; PRECISESADS Consortium: none; MEAl-R: none; LB: none; JM: none.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Patient consent for publication Not required.
Ethics approval An ethical protocol was prepared, consensus was reached across all partners, academic and industrial, translated into all participants’ languages and approved by each of the local ethical committees of the clinical recruitment centres. The studies adhered to the standards set by the International Conference on Harmonization and Good Clinical Practice (ICH-GCP), and to the ethical principles that have their origin in the Declaration of Helsinki (2013). The protection of the confidentiality of records that could identify the included subjects is ensured as defined by the EU Directive 2001/20/EC and the applicable national and international requirements relating to data protection in each participating country. The CS study is registered with number NCT02890121, and the inception study with number NCT02890134 in ClinicalTrials.gov. The study (PRECISESADS cross-sectional study) was approved by the following ethic committees: Comitato Etico Area 2 (Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico di Milano and University of Milan); approval no. 425bis 19 November 2014, and no. 671_2018 19 September 2018; Klinikum der Universitaet zu Koeln, Cologne, Germany. Geschaftsstelle Ethikkommission; Pôle de pathologies rhumatismales systémiques et inflammatoires, Institut de Recherche Expérimentale et Clinique, Université catholique de Louvain, Brussels, Belgium. Comité d Èthique Hospitalo-Facultaire; University of Szeged, Szeged, Hungary. Csongrad Megyei Kormanyhivatal; Hospital Clinic I Provicia, Institut d’Investigacions Biomèdiques August Pi i Sunyer, Barcelona, Spain. Comité de Ética de Investigación Clínica del Hospital Clínic de Barcelona. Hospital Clinic del Barcelona; Servicio Andaluz de Salud, Hospital Universitario Reina Sofía Córdoba, Spain. Comité de Ética de la Investigación de Centro de Granada (CEI – Granada); Centro Hospitalar do Porto, Portugal. Comissao de ética para a Saude – CES do CHP; Centre Hospitalier Universitaire de Brest, Hospital de la Cavale Blanche, Avenue Tanguy Prigent 29609, Brest, France. Comité de Protection des Personnes Ouest VI; Hôpitaux Universitaires de Genève, Switzerland. DEAS –Commission Cantonale d’éthique de la recherche Hôpitaux Universitaires de Genève; Biobanco del Sistema Sanitario Público de Andalucía, Granada, Spain; Katholieke Universiteit Leuven, Belgium. Commissie Medische Ethiek UZ KU Leuven /Onderzoek; Charite, Berlin, Germany. Ethikkommission; Medizinische Hochschule Hannover, Germany. Ethikkommission.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Summary statistics of the SSc meta-GWAS is available through the NHGRI-EBI GWAS Catalog (https://www.ebi.ac.uk/gwas/ downloads/summary-statistics) (‘Systemic Sclerosis’ and/or ‘Lopez-Isac/ Martin’ search terms). PRECISESADs data are available upon request at PRECISESADs consortium. All other data are contained in the article file and its supplementary information or available upon reasonable request to the corresponding authors.