Article Text

Abstract
Objective Tanezumab, a nerve growth factor inhibitor, was investigated for osteoarthritis (OA) of the hip or knee in a study with 24-week treatment and 24-week safety follow-up.
Methods This double-blind, randomised, phase III study enrolled adults in Europe and Japan with moderate-to-severe OA who had not responded to or could not tolerate standard-of-care analgesics. Patients were randomised to tanezumab 2.5 mg or 5 mg subcutaneously or matching placebo every 8 weeks (three doses). Co-primary end points were change from baseline to week 24 in Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC) Pain and Physical Function, and Patient’s Global Assessment of OA (PGA-OA). Joint safety and neurological assessments continued throughout the 48-week study.
Results From March 2016 to December 2017, 849 patients were randomised and evaluated (placebo n=282, tanezumab 2.5 mg n=283, tanezumab 5 mg n=284). At week 24, there was a statistically significant improvement from baseline for tanezumab 5 mg compared with placebo for WOMAC Pain (least squares mean difference±SE –0.62±0.18, p=0.0006), WOMAC Physical Function (–0.71±0.17, p<0.0001) and PGA-OA (–0.19±0.07, p=0.0051). For tanezumab 2.5 mg, there was a statistically significant improvement in WOMAC Pain and Physical Function, but not PGA-OA. Rapidly progressive osteoarthritis (RPOA) was observed in 1.4% (4/283) and 2.8% (8/284) of patients in the tanezumab 2.5 mg and tanezumab 5 mg groups, respectively and none receiving placebo. Total joint replacements (TJRs) were similarly distributed across all three treatment groups (6.7%–7.8%). Tanezumab-treated patients experienced more paraesthesia (5 mg) and hypoaesthesia (both doses) than placebo.
Conclusion Tanezumab 5 mg statistically significantly improved pain, physical function and PGA-OA, but tanezumab 2.5 mg only achieved two co-primary end points. RPOA occurred more frequently with tanezumab 5 mg than tanezumab 2.5 mg. TJRs were similarly distributed across all three groups.
Trial registration number NCT02709486.
- osteoarthritis
- knee osteoarthritis
- analgesics
Data availability statement
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual deidentified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programmes that have been terminated (ie, development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: https://creativecommons.org/licenses/by-nc-nd/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
A systematic review and meta-analysis of the efficacy and safety of tanezumab for osteoarthritis (OA), published in 2015, was based on a search of PubMed and Embase using relevant keywords (tanezumab, fulranumab, fasinumab, anti-nerve growth factor (NGF), NGF, osteoarthritis or OA) and lists of studies obtained from the developers.
The authors imposed no eligibility restrictions on the age of participants, the duration of disease or concomitant medication use (non-steroidal anti-inflammatory drug or opioids), nor language or publication date/status restrictions.
This systematic review showed that the early tanezumab studies investigated mostly intravenous administration with primary end points evaluated at 16 weeks.
Doses were body weight-adjusted or administered as fixed doses (up to tanezumab 10 mg).
The few studies performed using subcutaneous injection were impacted by temporary partial clinical holds placed on non-cancer pain studies of all NGF monoclonal antibodies following joint safety findings in clinical trials, which were adjudicated as rapidly progressive osteoarthritis, and concerns regarding histomorphological changes in the sympathetic nervous system reported in preclinical studies.
After a comprehensive assessment of the clinical data related to joint safety and neurological safety as well as additional preclinical studies of sympathetic nervous system morphology and function, a risk minimisation plan including comprehensive surveillance for joint safety and sympathetic neurological safety was implemented.
The phase III OA programme conducted post-2015 used only lower doses of tanezumab administered subcutaneously in difficult-to-treat patients, and incorporated extensive risk management procedures to maintain patient safety.
Key messages
What does this study add?
Of the phase III OA programme, this is the first study to assess the efficacy of tanezumab 2.5 mg and 5 mg every 8 weeks for 24 weeks with additional 24-week safety follow-up in patients with moderate-to-severe OA and inadequate response to standard-of-care analgesics.
How might this impact on clinical practice or future developments?
This study demonstrates the efficacy of subcutaneous tanezumab in difficult-to-treat patients with OA.
Adjudicated joint safety end points occurred only in tanezumab-treated patients.
Total joint replacements were similarly distributed across all three treatment groups.
A longer-term active-controlled study (ClinicalTrials.gov, NCT02528188) will provide data to further characterise the risk-benefit of tanezumab in patients with OA.
Introduction
Globally, osteoarthritis (OA) is a leading cause of disability1 with joint pain that often makes common daily activities difficult. Patients with knee or hip OA also have a higher risk of mortality than the general population due to increased cardiovascular events.2
To manage the pain associated with OA, guidelines recommend a combination of non-pharmacological approaches and analgesics3 4 including acetaminophen (paracetamol), non-steroidal anti-inflammatory drugs (NSAIDs) and opioids. Limited efficacy in some patients and concerns about adverse effects5 6 mean that an alternative to current pharmacological treatment options is needed.
Nerve growth factor (NGF) is involved in pain signalling7 and has been implicated in OA pain.8 Early clinical trials of the NGF monoclonal antibody tanezumab investigated mostly intravenous administration of doses that were body weight-adjusted or administered as a fixed dose (up to 10 mg), and used primary efficacy end points measured at 16 weeks or earlier.9 Cases of rapidly progressive osteoarthritis (RPOA) were observed in some of these studies.10 Due to these joint safety concerns and histomorphological changes in the sympathetic nervous system observed in preclinical animal studies,11 the US Food and Drug Administration placed partial clinical holds on studies of NGF antibodies in 2010 and 2012, respectively. Investigation of these events provided evidence that led to the subsequent lifting of the tanezumab clinical holds, and an overall risk minimisation strategy including comprehensive monitoring of joint and neurological safety was implemented in subsequent studies. The phase III OA programme conducted after the clinical holds were removed (post-2015) used only lower doses of tanezumab administered subcutaneously in difficult-to-treat patients, incorporated extensive risk mitigation and surveillance, excluded patients with evidence of or risk factors for RPOA or who were unsuitable for joint replacement and restricted chronic concomitant use of NSAIDs while in the study.
The current study was designed to assess the efficacy (24 weeks) and safety (total 48 weeks, including a 24-week post-treatment safety follow-up period) of subcutaneous tanezumab (2.5 mg or 5 mg vs placebo, every 8 weeks for a total of three doses) in patients with moderate-to-severe OA with an inadequate response to, or who could not tolerate, standard-of-care analgesics including acetaminophen, NSAIDs and tramadol or opioids.
Methods
Study design
This was a phase III, randomised, double-blind, placebo-controlled, parallel-group, multicentre trial conducted at 104 hospital, clinical research or general practice sites in Europe and Japan from March 2016 to November 2018. Patients were recruited directly by investigators, through referrals, and also by advertising.
The study comprised a screening period (up to 37 days prior to randomisation), a 24-week double-blind treatment period and a 24-week safety follow-up period (figure 1). The screening period included a washout phase (lasting a minimum of 2 days for all prohibited pain medications), if required, and an initial pain assessment period (the 7 days prior to randomisation/baseline) to establish average pain levels in the index joint.

Study design. Scheduled in-clinic visits occurred at screening, baseline and weeks 2, 4, 8, 12, 16, 24, 32 and 48, with telephone contact scheduled for weeks 20, 28, 36, 40 and 44. Patients who did not complete the double-blind treatment period were still followed through the 24-week safety follow-up period.
Patients
Adults (≥18 years of age, male or female) were eligible for inclusion if they had a diagnosis of OA of the hip or knee in the index joint based on American College of Rheumatology criteria12 13 confirmed radiographically (Kellgren-Lawrence14 grade ≥2) as diagnosed by the Central Readers. They were also required to have a documented history of: acetaminophen providing insufficient pain relief; and oral NSAIDs providing inadequate pain relief or an inability to be taken due to intolerance or contraindication; and inadequate pain relief from, or intolerance or contraindication to, either tramadol or opioids (or unwillingness to take opioids). Other key inclusion criteria were Western Ontario and McMaster Universities Osteoarthritis Index (WOMAC15) Pain subscale score ≥5 in the index joint at screening and baseline, WOMAC Physical Function subscale score ≥5 in the index joint at baseline (all as single time points, and an increase after washout of analgesics was not required); and ‘fair’, ‘poor’ or ‘very poor’ Patient’s Global Assessment of OA (PGA-OA) at baseline.
Key exclusion criteria were radiographic evidence of specified bone or joint conditions (RPOA, atrophic or hypotrophic OA, subchondral insufficiency fracture, spontaneous osteonecrosis of the knee, osteonecrosis or pathological fracture) or other conditions (excessive malalignment of the knee, severe chondrocalcinosis, other arthropathies [eg, rheumatoid arthritis], systemic metabolic bone disease, large cystic lesions, primary or metastatic tumour lesions, stress or traumatic fracture) identified by a group of centralised musculoskeletal radiologist readers.16 Also excluded were those with a history of osteonecrosis or osteoporotic fracture, significant trauma or surgery to a knee, hip or shoulder within the previous year. Patients with a history of carpal tunnel syndrome with signs or symptoms in the 1 year prior to screening were excluded, as were those with a history or presence of clinically significant neurological disease or psychiatric disorder. Use of an oral or intramuscular corticosteroid within the last 30 days, intra-articular corticosteroid injection in the index joint within 12 weeks or in any other joint within the last 30 days, intra-articular hyaluronic acid injection in the index joint within 30 days or long-acting hyaluronic acid formulation injection in the index joint within 18 weeks also resulted in exclusion.
Randomisation and blinding
Patients were randomised in a 1:1:1 ratio, stratified by index joint (hip or knee) and most severe Kellgren-Lawrence grade (2, 3 or 4) in any hip or knee, to one of three parallel groups: tanezumab 2.5 mg, tanezumab 5 mg or matching placebo.
A code was created by the computerised randomisation schedule, which was generated and held by a person independent of the study. All patients, investigators, study coordinators, clinical site staff, clinical research associates and the sponsor’s staff directly involved in the study were blinded.
Procedures
All study treatments (placebo, tanezumab 2.5 mg and tanezumab 5 mg) were provided by Pfizer in matching glass prefilled syringes. Treatments were administered subcutaneously (1 mL) into the abdomen or anterior thigh by study site staff at baseline, week 8 and week 16.
Use of non-study analgesic medication was restricted through week 32, as detailed below. After week 32, patients could be started on any standard treatment for OA pain.
Except for the 24 hours prior to any study visit for efficacy assessments, acetaminophen (rescue therapy, up to 4000 mg/day or as permitted by local or national labelling) was allowed during the initial pain assessment period and up to 5 days/week up to week 24, and then as needed until week 32. Limited use of NSAIDs (prescription or over-the-counter, ≤10 days per 8-week period, total ≤40 days) was permitted on an occasional basis until week 32 for self-limiting conditions not related to OA, but not within 48 hours or five half-lives, whichever was greater, of study visits for efficacy assessments. All other analgesics were prohibited from 48 hours prior to the screening initial pain assessment period until week 32.
Efficacy outcomes
The three co-primary efficacy end points were change from baseline to week 24 in WOMAC15 Pain subscale score, WOMAC Physical Function subscale score and PGA-OA score. WOMAC Pain and Physical Function subscale scores assessed symptoms over the previous 48 hours in the index joint on an 11-point numeric rating scale (NRS) and PGA-OA was rated on a 5-point Likert scale. An electronic device was used during clinic visits to complete the study questionnaires, including WOMAC and PGA-OA.
Key secondary end points included the proportion of patients achieving at least 50% reduction in WOMAC Pain subscale score at week 24, change from baseline to week 2 in WOMAC Pain subscale score and change from baseline to week 1 in the average pain score in the index joint. Patients used an electronic diary to rate their average pain in the index joint over the previous 24 hours on an 11-point NRS (from 0=no pain to 10=worst possible pain). Other secondary end points included the proportion of patients achieving at least 30%, 70% and 90% reductions in WOMAC Pain subscale score at week 24.
Safety outcomes
Safety assessments included treatment-emergent adverse events (AEs), physical examination, laboratory tests, vital signs and orthostatic blood pressure, 12-lead electrocardiograms and antitanezumab antibody assessments. AEs were coded using Medical Dictionary for Regulatory Activities V.21.1 and severity and causality were assessed by study investigators. AEs were considered serious based on established definitions (those that were fatal or life-threatening, required new or extended hospitalisation or resulted in significant or persistent disability, or birth defects).17
Joint safety monitoring included radiography of bilateral hips, knees and shoulders at screening, week 24 and week 48, evaluated by a Central Reader. After the baseline visit, the Central Reader requested follow-up MRI if there were equivocal radiographic findings that required investigation. Musculoskeletal examinations, evaluation of relevant AEs and monitoring of pain scores to detect increased, severe, persistent joint pain occurred throughout the study. Investigators could request imaging, including MRI, to evaluate patients who reported increased, severe, persistent pain. Those images were also evaluated by the Central Reader. All possible or probable joint safety events identified by the Central Reader or the investigator, and all total joint replacements (TJRs), for any reason, were reviewed by a blinded Adjudication Committee (comprised of external experts in orthopaedic surgery, orthopaedic pathology, rheumatology and musculoskeletal radiology).
Joint safety was evaluated based on the numbers of patients with adjudicated joint safety end points (RPOA type 1 or type 2, subchondral insufficiency fracture, primary osteonecrosis or pathological fracture) and TJRs. RPOA type 1 was defined as a significant loss of joint space width ≥2 mm (predicated on optimal joint positioning) within approximately 1 year, without gross structural failure; RPOA type 2 was defined as abnormal bone loss or destruction, including limited or total collapse of at least one subchondral surface, that is not normally present in conventional end-stage OA.18
Neurological assessment included the Neuropathy Impairment Score19 and Survey of Autonomic Symptoms.20 Patients were evaluated by a consulting neurologist if an AE of peripheral neuropathy or abnormal peripheral sensation was reported as serious or severe, caused study withdrawal or was unresolved at the end of study participation; or if there was a clinically significant neurological examination abnormality which met one or more of the criteria noted above. Patients with AEs of any seriousness or severity that were suggestive of sympathetic autonomic neuropathy (ie, bradycardia, orthostatic hypotension, syncope, anhidrosis or hypohidrosis) were evaluated by a cardiologist or neurologist, as specified in the protocol.
Statistical analysis
From two previous clinical trials (ClinicalTrials.gov, NCT0073390221 and NCT0074447122), treatment differences between tanezumab 2.5 mg/tanezumab 5 mg and placebo were assumed to be –0.75/–0.77 for WOMAC Pain, –1.01/–1.05 for WOMAC Physical Function and –0.25/–0.34 for PGA-OA, and within-group SD were 2.76, 2.63 and 0.94 for the three outcomes, respectively. A sample size of 270 patients per treatment group was estimated to provide ~80% power to achieve statistical significance at the 5% two-sided level for the two comparisons of tanezumab (2.5 mg and 5 mg) versus placebo over all three co-primary end points.
The primary efficacy population comprised all those randomised who received ≥1 dose of study treatment. Safety was also assessed in this population.
The graphical approach of gatekeeping strategy23 was applied for co-primary and key secondary end points to control the family-wise type 1 error rate of 5% (two-sided). The co-primary efficacy end points were analysed using analysis of covariance (ANCOVA), with model terms for baseline score of the corresponding end point, baseline diary average pain, index joint (knee or hip), highest Kellgren-Lawrence grade and treatment group, with study site as a random effect. Missing data at week 24 were handled with a multiple imputation strategy dependent on the reason for discontinuation. Based on the gatekeeping strategy, co-primary end points of tanezumab 5 mg were first tested versus placebo and, if statistically significant (p≤0.05), co-primary end points of tanezumab 2.5 mg were then tested versus placebo. A tanezumab treatment group was considered superior to placebo only if all three co-primary end points were statistically significant. Gated by the primary analysis (ie, conducted if the co-primary end points were statistically significant for both treatment groups), the key secondary end points were tested using logistic regression (WOMAC Pain responders) and ANCOVA (WOMAC Pain at week 2 and average pain at week 1).
Unblinded safety data were reviewed regularly throughout by an independent, external Data Monitoring Committee.
SAS software V.9.4 (SAS Institute, Cary, North Carolina, USA) was used for all statistical analyses. The study was registered on ClinicalTrials.gov (NCT02709486).
Patient and public involvement
This research was done without patient involvement in the design of the study: the research question was derived from the tanezumab phase III development programme, as agreed with regulatory agencies. The study design was based on earlier studies, using outcome measures that had been developed/validated academically with patient participation. The patients were recruited directly by investigators, through referrals and also by advertising, and gave their written informed consent after explanation of the details of the trial. They were not asked to assess the burden of the intervention or time required. The research findings will be disseminated via the standard channels, including a plain language summary.
Results
Of the 2145 patients who were screened, 849 were randomised between 29 March 2016 and 21 December 2017 and all of them received study medication; 58.2% (1248/2145) of patients screened did not meet the study entry criteria, mostly due to insufficient pain or not meeting the radiographic criteria (figure 2). During the double-blind treatment period, most patients received all three planned doses of study medication (85.8% for placebo, 93.3% for tanezumab 2.5 mg and 93.0% for tanezumab 5 mg). The double-blind 24-week treatment period was completed by 750 (88.3%) patients, 696 (82.0%) completed both the treatment and safety follow-up periods and 726 (85.5%) completed the 24-week safety follow-up period. All 849 patients contributed to both efficacy and safety datasets (figure 2).

Patient disposition. *Patients screened but not randomised for a reason not related to a specific eligibility criterion. †After either completing the treatment period or discontinuing the treatment period.
Demographics and baseline characteristics were comparable across treatment groups (table 1). The age of the population ranged from 26 to 89 years and the majority (587/849, 69.1%) were female; for 83.0% (705/849) of the patients, their OA index joint was a knee.
Demographics and baseline characteristics
At week 24, there was a statistically significant improvement from baseline in all three co-primary end points for patients receiving tanezumab 5 mg compared with placebo: WOMAC Pain (least squares (LS) mean difference±SE–0.62±0.18, p=0.0006), WOMAC Physical Function (–0.71±0.17, p<0.0001) and PGA-OA (–0.19±0.07, p=0.0051) (figure 3). Tanezumab 2.5 mg was not statistically significantly better than placebo for all three co-primary end points: at week 24 there was a statistically significant improvement for WOMAC Pain (–0.46±0.18, p=0.0088) and Physical Function (–0.59±0.18, p=0.0008), but PGA-OA (–0.11±0.07, p=0.1092) was not significantly different from placebo (figure 3).

Change from baseline to week 24 in the co-primary end points: WOMAC Pain subscale score, WOMAC Physical Function subscale score and PGA-OA score. *p≤0.05; **p≤0.01; ***p≤0.001 vs placebo. WOMAC Pain assessed pain over the previous 48 hours in the index joint, and was the mean of five questions each scored on an 11-point numerical rating scale (NRS), from 0 to 10 with higher score indicating more pain. WOMAC Physical Function assessed ability to move around and perform activities of daily living over the previous 48 hours in the index joint, and was the mean of 17 questions each scored on an 11-point NRS, from 0 to 10 with higher score indicating worse function. PGA-OA was rated on a 5-point Likert scale from 1=‘very good’ (asymptomatic and no limitation of normal activities) to 5=‘very poor’ (very severe symptoms which are intolerable and inability to carry out all normal activities) in answer to the question “Considering all the ways your osteoarthritis in your hip/knee affects you, how are you doing today?” Reproduced from Berenbaum F, Blanco F, Guermazi A, Vignon E, Miki K, Yamabe T, Viktrup L, Junor R, Carey W, Brown M, Verburg K, West C. Ann Rheum Dis. 2019;78(suppl 2):262–263, with permission from BMJ Publishing Group Ltd. LS, least squares; PGA-OA, Patient’s Global Assessment of osteoarthritis; WOMAC, Western Ontario and McMaster Universities Osteoarthritis Index.
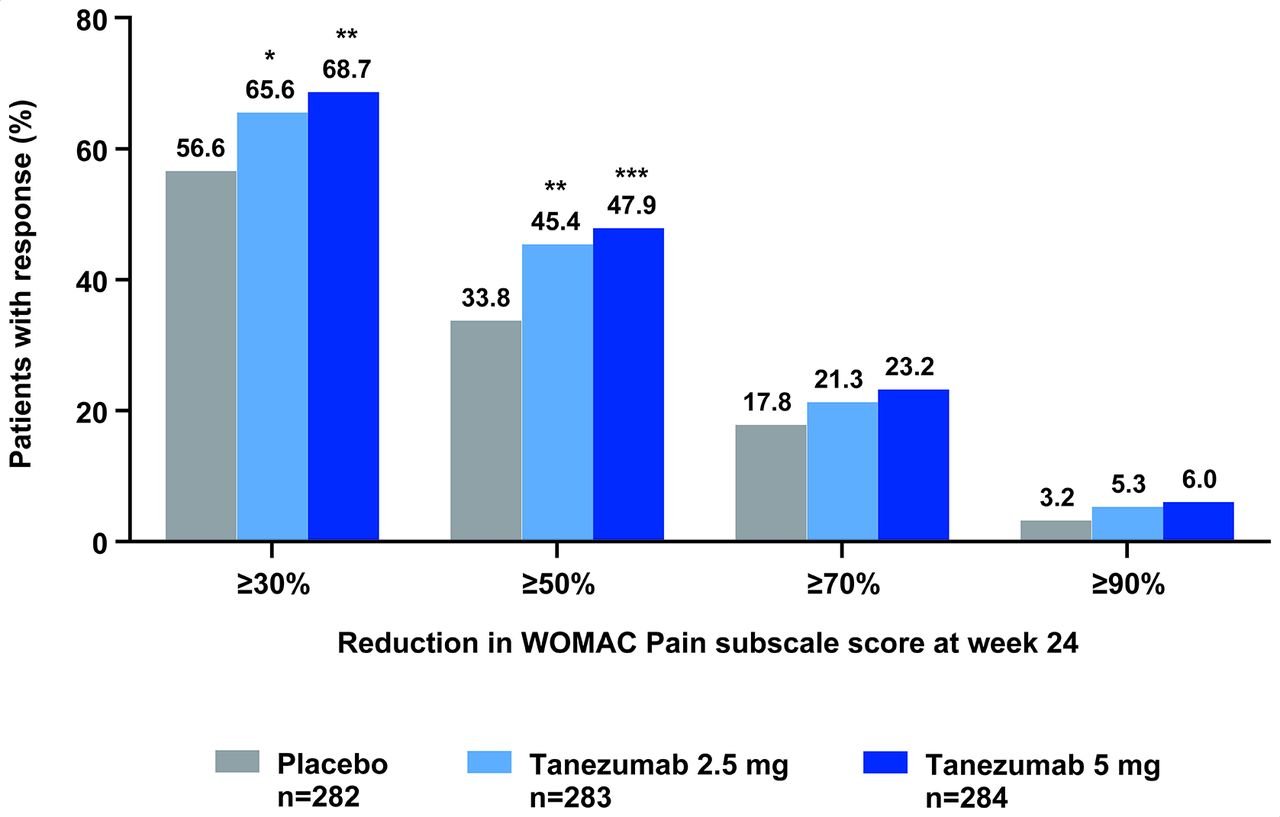
From the predefined gatekeeping strategy, because the PGA-OA end point was not met for the tanezumab 2.5 mg treatment group, further hypothesis testing of the three key secondary end points for both tanezumab treatment groups could not be performed. Without adjustment for multiple comparisons, the three key secondary efficacy end points for both the tanezumab 5 mg and tanezumab 2.5 mg treatment groups were better than the placebo treatment group (all nominal p≤0.05) (table 2). The proportion of patients achieving at least a 50% reduction from baseline in WOMAC Pain subscale score at week 24 was 33.8% (placebo), 45.4% (tanezumab 2.5 mg) and 47.9% (tanezumab 5 mg), with a similar pattern for the other categories of response (≥30%, ≥70%, ≥90%) (figure 4).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Proportion of patients achieving at least 30%, 50%, 70% and 90% reductions in WOMAC Pain subscale score at week 24. *P≤0.05; **p≤0.01; ***p≤0.001 vs placebo. Mixed baseline/last observation carried forward. Nominal, unadjusted p value. In line with the predefined gatekeeping strategy, hypothesis testing of the three key secondary end points could not be performed: no key secondary end points can be declared as statistically significantly better than placebo treatment. WOMAC, Western Ontario and McMaster Universities Osteoarthritis Index.
Results for key secondary end points
During the double-blind treatment period, a total of 55.0%, 53.0% and 57.0% of patients in the placebo, tanezumab 2.5 mg and tanezumab 5 mg groups, respectively, experienced AEs (table 3). During this period, arthralgia was the most common AE in the placebo group, and of AEs that occurred in ≥3% of patients in any of the groups, back pain and OA were more frequent in both tanezumab treatment groups than placebo (table 3). Study discontinuations due to AEs were similar across the three groups (table 3). Serious AEs occurred in 1.1% (placebo), 2.8% (tanezumab 2.5 mg) and 3.2% (tanezumab 5 mg) of patients during the double-blind treatment period (table 3), and none was considered treatment-related by investigators. There were two deaths during the double-blind treatment period in the tanezumab 5 mg group, and neither were considered treatment-related (table 3).
Summary of treatment-emergent adverse events (AEs) reported by the investigator
Of the 79 patients requiring adjudication for joint safety, most (58/79, 73.4%) were adjudicated as normal progression of OA (table 4). A total of 14 patients had an adjudicated joint safety end point, all receiving tanezumab, including 1.4% (4/283) and 2.8% (8/284) adjudicated as RPOA in the tanezumab 2.5 mg and tanezumab 5 mg groups, respectively (table 4).
Summary of adjudicated joint safety findings (baseline to week 48)
A total of eight joints in eight patients were adjudicated as RPOA type 1, of which three of eight were in the tanezumab 2.5 mg group: these included two knees (baseline Kellgren-Lawrence grades 0 and 1, both non-index joints) and one index hip (baseline Kellgren-Lawrence grade 3). The other five joints with RPOA type 1 were in the tanezumab 5 mg group: these were all knees of baseline Kellgren-Lawrence grade 2 (four joints, of which three were index joints and one was a non-index joint) or 3 (one non-index joint). Two of the eight joints affected by RPOA type 1 underwent TJR (both baseline Kellgren-Lawrence grade 3, of which one was the index hip in the tanezumab 2.5 mg group and one a non-index knee in the tanezumab 5 mg group).
A total of four joints in four patients were adjudicated as RPOA type 2, of which one of four was in the tanezumab 2.5 mg group, and was an index hip (baseline Kellgren-Lawrence grade 4) which underwent TJR. The other three joints with RPOA type 2 were in the tanezumab 5 mg group: all were hips (one index and two non-index) of baseline Kellgren-Lawrence grade 3, of which one (the index joint) underwent TJR.
One patient in the tanezumab 5 mg group had a joint adjudicated as primary osteonecrosis, a non-index hip with baseline Kellgren-Lawrence grade 0 that did not undergo TJR. One patient in the tanezumab 2.5 mg group had a joint adjudicated as subchondral insufficiency fracture, a non-index knee with baseline Kellgren-Lawrence grade 2 that did not undergo TJR.
Overall, there were 63 TJRs in 61 patients, evenly distributed across the three treatment groups with the majority (54/63, 85.7%) being index joints (table 4). In most cases, the joint replacement surgery was considered to be elective (40/63, 63.5%), that is, not associated with an AE or an adjudicated joint safety end point. Four patients (4/61, 6.6%) had a TJR during the treatment period (0, two and two patients in the placebo, tanezumab 2.5 mg and tanezumab 5 mg treatment groups, respectively), 52 patients (52/61, 85.2%) after completing treatment but during, or after completing or discontinuing the safety follow-up period (15, 19 and 18 patients in the placebo, tanezumab 2.5 mg and tanezumab 5 mg treatment groups, respectively) and five patients (5/61, 8.2%) after discontinuing treatment and continuing or not in the safety follow-up period (four, one and 0 patients in the placebo, tanezumab 2.5 mg and tanezumab 5 mg treatment groups, respectively). Two of the patients, both in the placebo group, underwent TJR in a second joint.
During the double-blind treatment period, paraesthesia and hypoaesthesia were more frequent with tanezumab 5 mg than placebo, and hypoaesthesia was also more frequent with tanezumab 2.5 mg than placebo (table 3). No patients had severe paraesthesia or hypoaesthesia; one patient with mild hypoaesthesia discontinued treatment in the tanezumab 5 mg group and had a final diagnosis of carpal tunnel syndrome (table 3). During the treatment period, carpal tunnel syndrome was observed in three patients, including two in the tanezumab 2.5 mg group and one in the tanezumab 5 mg group. A total of 22 patients reported an AE which required a neurological consultation. Following review of the clinical information for these patients, a blinded external neurologist reported primary diagnoses of radiculopathy (one, five and three patients in the placebo, tanezumab 2.5 mg and tanezumab 5 mg groups, respectively), mononeuropathy (one, five and three patients, respectively) and polyneuropathy (one patient in the tanezumab 2.5 mg group), with other patients having no neuropathic symptoms or signs (one patient in the placebo group) or neuropathic symptoms but no clinically significant signs (one patient each in the placebo and tanezumab 2.5 mg groups).
AEs potentially related to sympathetic nervous system function occurred with low frequencies in all treatment groups during the double-blind treatment period, including bradycardia (two, two and four patients in the placebo, tanezumab 2.5 mg and tanezumab 5 mg groups, respectively), orthostatic hypotension (0, 0 and three patients, respectively) and syncope (0, 0 and one patient, respectively). Based on protocol-specified cardiology or neurology consultations for patients reported to have AEs potentially related to sympathetic nervous system function, study investigators determined that no patient in any treatment group was considered to have a sympathetic neuropathy.
Discussion
This study showed that in patients who have not responded to or could not tolerate standard-of-care analgesics, tanezumab 5 mg, administered subcutaneously, statistically significantly improved all three co-primary efficacy end points at 24 weeks: WOMAC Pain, WOMAC Physical Function and PGA-OA. Tanezumab 2.5 mg improved WOMAC Pain and WOMAC Physical Function, but was not statistically significant on the PGA-OA end point at week 24. The proportion of patients achieving at least a 50% reduction from baseline in WOMAC Pain at week 24 was 33.8% for placebo, compared with 45.4% for tanezumab 2.5 mg and 47.9% for tanezumab 5 mg (figure 4) and rates of discontinuation due to insufficient clinical response were low (figure 2). Tanezumab was generally well tolerated through the 24-week treatment and 24-week safety follow-up periods, with similarly low rates of discontinuations due to AEs observed among patients receiving tanezumab and placebo. TJRs were similarly distributed across all three treatment groups. Adjudicated joint safety end points occurred only in tanezumab-treated patients, and more frequently with tanezumab 5 mg than tanezumab 2.5 mg.
The efficacy observed in the current study is supported by two previous placebo-controlled studies in patients with knee21 or hip22 OA that administered tanezumab 2.5 mg and 5 mg intravenously, with both doses resulting in improvement across most of the end points at week 24 (online supplementary tables 1 and 2: some of the 24-week data are previously unpublished). As with the current study, significant improvements in WOMAC Pain and Physical Function were seen with tanezumab 2.5 mg at week 24 in both those intravenous studies. The tanezumab 5 mg dose did not reach significance on WOMAC Pain or the PGA-OA at week 24 in the intravenous knee study (online supplementary table 1). The placebo response for PGA-OA at week 24 was larger in the current study (–0.72, figure 3) than that observed in these earlier intravenous studies (–0.33 to –0.52, online supplementary tables 1 and 2).
Supplemental material
The large placebo response on the PGA-OA end point in the current study may potentially have contributed to the failure of the tanezumab 2.5 mg dose to reach statistical significance on this end point. Contextual factors can contribute considerably to the treatment effect in OA,24 with multiple factors influencing the size of the placebo response,25 and it is possible that treatment expectations were high in the current difficult-to-treat population receiving subcutaneous injection of a medication previously shown to be effective. A reduced treatment response to tanezumab or an administration route effect cannot be excluded, but there were also differences in patient populations. It should also be noted that some pre-hold OA studies (conducted before 2015) had a flare design. The current study was conducted in Europe and Japan in patients with moderate-to-severe OA for whom standard-of-care treatment was inadequate or unsuitable. Baseline WOMAC Pain scores were less severe in the current population compared with the previous intravenous tanezumab knee21 and hip22 OA studies, although baseline joint radiographic findings were more severe, with 36% of patients with Kellgren-Lawrence grade 4 in the current study compared with <21% in the previous studies.21 22 The association between pain and radiographic findings has not always been consistent,26 but a relationship has been reported for knee OA.27 Overall, the proportion of patients with a moderate (≥30%) or substantial (≥50%) clinically important improvement in WOMAC Pain28 with tanezumab compared with placebo at 24 weeks was statistically significant in the current study, although smaller than observed in the intravenous tanezumab knee21 and hip22 studies (online supplementary tables 1 and 2). Notwithstanding the limitation in dichotomising continuous data,29–32 an effect size (placebo-adjusted LS mean change divided by model-based SD) of 0.20 has been proposed as the lower bound for a small meaningful effect (or change in pain).33 In our study, the effect size (placebo-adjusted) for tanezumab 2.5 mg and 5 mg dose regimens at week 24 was modest at 0.24 (0.46/1.93=0.24) and 0.32 (0.62/1.93=0.32), respectively, although both above the suggested lower threshold for meaningfulness.
In the current study, tanezumab was generally well tolerated during the 24-week treatment period, with similarly low rates of discontinuation due to AEs observed among patients taking tanezumab and placebo. The observed AEs were consistent with previous studies,9 21 22 34 35 with no new safety signals identified. Paraesthesia was more frequent with tanezumab 5 mg than placebo, and hypoaesthesia was more frequent with both tanezumab doses than placebo. TJRs were similarly distributed across all three treatment groups, mostly in joints with a baseline Kellgren-Lawrence grade of 3 or 4. Non-elective joint replacements were more frequent in the tanezumab 5 mg treatment group (11/20 joints, 55.0%) compared with tanezumab 2.5 mg (6/22 joints, 27.3%) or placebo (6/21 joints, 28.6%). Adjudicated joint safety end points were only reported in tanezumab-treated patients, and there were twice as many cases of RPOA type 1 (n=8) than RPOA type 2 (n=4) (table 4). The tanezumab 5 mg treatment group had the highest incidence of RPOA type 1, RPOA type 2 and primary osteonecrosis (one case), whereas the only subchondral insufficiency fracture was observed in the tanezumab 2.5 mg group. OA is frequently a multijoint disease, and the joints adjudicated as RPOA included both index and non-index joints. All four joints adjudicated as RPOA type 2 in the tanezumab treatment groups had a baseline Kellgren-Lawrence grade of 3 or 4. There were three joints with baseline Kellgren-Lawrence grade 0 or 1, which were adjudicated as RPOA type 1 (two joints in the tanezumab 2.5 mg treatment group) or osteonecrosis (one joint in the tanezumab 5 mg treatment group).
The frequency of RPOA in the current study (12/567, 2.1% of tanezumab-treated patients) is higher than that seen in a similarly designed but shorter 16-week North American study with less exposure to the 5 mg dose (6/464, 1.3%) and fewer patients with Kellgren-Lawrence grade 4 OA.34 Comparison of data from the current study with data from previous tanezumab studies (conducted before 2015) that did not include risk mitigation measures and enhanced surveillance for joint safety events is difficult.
The comprehensive joint safety assessment used in the current tanezumab development programme was strengthened after the clinical holds were removed and provides radiographic and symptomatic details on the natural progression of OA in patients with or without treatment. The sequential radiography allows for identification of RPOA type 1 that was not done before, and the Central Reader and Adjudication Committee evaluation of imaging allow for a more precise identification of RPOA type 2 cases. These features may help to identify a potential correlation between RPOA type 1 and type 2 and better characterise the risk-benefit of tanezumab in patients with OA. As the pathophysiology of RPOA is poorly understood,10 the tanezumab clinical programme represents the largest dataset available to evaluate RPOA.
The strengths of this study compared with the pre-hold programme (studies conducted before 2015) include the recruitment of difficult-to-treat patients and comprehensive joint safety and sympathetic neurological surveillance, and 24-week safety follow-up. The limitations include a relatively high rate of screening failures. Understanding of the pathogenesis of joint safety events seen with tanezumab, and how this is similar to or different from the rapid joint space narrowing or rapidly destructive arthropathies seen elsewhere, is currently limited. A longer-term active-controlled study (NCT02528188) will provide more data to further characterise the risk-benefit of tanezumab in patients with OA.
Conclusion
This study demonstrates subcutaneous tanezumab at a dose of 5 mg every 8 weeks statistically significantly improves pain, physical function and PGA-OA at 24 weeks in patients with moderate-to-severe OA who have not responded to or could not tolerate standard-of-care analgesics. Tanezumab 2.5 mg improved pain and function in patients with moderate-to-severe OA, but did not reach statistical significance on the other co-primary end point, PGA-OA. Adjudicated joint safety end points occurred only in tanezumab-treated patients, with RPOA in 1.4% and 2.8% of patients in the tanezumab 2.5 mg and tanezumab 5 mg groups, respectively, and in none receiving placebo. The frequency of TJR was similar across all three treatment groups.
Data availability statement
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual deidentified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programmes that have been terminated (ie, development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The deidentified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Ethics statements
Patient consent for publication
Ethics approval
The study protocol was approved by the appropriate Institutional Review Board or Independent Ethics Committee at each participating investigational centre, and all patients provided written informed consent prior to entering the study. The study was conducted in compliance with the Declaration of Helsinki and International Conference on Harmonisation Good Clinical Practice Guidelines.
Acknowledgments
The authors would like to thank all of the patients, investigators and study site personnel for participating in the tanezumab clinical development programme. The authors would also like to thank the Central Readers, the Data Monitoring Committee and the joint safety Adjudication Committee members. Some of these data were presented at the European Congress of Rheumatology (EULAR), Madrid, Spain, 12–15 June 2019 (Ann Rheum Dis 2019;78 (suppl 2):262–4).
References
Footnotes
Handling editor Josef S Smolen
Twitter @larhumato
Contributors AG, LV, KMV, MTB and CRW contributed to the conception or design of the study. All authors contributed to the acquisition, analysis or interpretation of data. TY was the study statistician. All authors contributed to drafting the manuscript and revising it critically for important intellectual content. All authors approved the final version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Funding The study was sponsored by Pfizer and Eli Lilly and Company. Medical writing support was provided by Kim Russell, PhD, of Engage Scientific Solutions (Horsham, UK) and was funded by Pfizer and Eli Lilly and Company. Pfizer is the manufacturer of tanezumab, which is being investigated for the treatment of patients with chronic pain. Manuscript authors from Pfizer contributed to the study design; data collection, management and interpretation of data; and the preparation, review and approval of the manuscript. Manuscript authors from Eli Lilly and Company contributed to the study design, interpretation of data and the preparation, review and approval of the manuscript.
Competing interests FB reports personal fees from Boehringer, Bone Therapeutics, Expanscience, Galapagos, Gilead, GSK, Merck Sereno, MSD, Nordic, Novartis, Pfizer, Regulaxis, Roche, Sandoz, Sanofi, Servier, UCB, Peptinov, TRB Chemedica, 4P Pharma, outside the submitted work. FJB reports grants and personal fees from Pfizer, and grants from AbbVie, UCB, Bristol-Meyers Squibb, Roche, Servier, Bioiberica, Sanofi, Grunenthal, GlaxoSmithKline, Lilly, Janssen, Regeneron, Amgen and TRB Chemedica, outside the submitted work. AG reports personal fees from Merck Serono, Pfizer, TissueGene, Roche, Galapagos, AstraZeneca, personal fees from Boston Imaging Core Lab, LLC, outside the submitted work. KM reports personal fees from Merck, Pfizer, Lilly, Ayumi, Mundi Pharma, Janssen, Nippon Zok, outside the submitted work. LV reports personal fees and other from Eli Lilly and Company, outside the submitted work. TY reports personal fees and other from Pfizer outside the submitted work. RJ reports personal fees and other from Pfizer outside the submitted work. WC reports personal fees and other from Pfizer outside the submitted work. MTB reports personal fees and other from Pfizer outside the submitted work, and has a patent pending (Tanezumab Method of Treatment). CRW reports personal fees and other from Pfizer outside the submitted work, and has a patent pending (Tanezumab Method of Treatment). KMV reports personal fees and other from Pfizer outside the submitted work.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.