Article Text

Abstract
Objectives SPIRIT head-to-head (H2H) is a 52-week (Wk) trial comparing ixekizumab (IXE) with adalimumab (ADA) for simultaneous American College of Rheumatology (ACR)50 and Psoriasis Area and Severity Index (PASI)100 responses in 566 patients (distributed evenly across both groups) with psoriatic arthritis (PsA). IXE was superior to ADA for this primary end point at Wk24. We aimed to determine the final efficacy and safety results through Wk52 including a prespecified subgroup analysis of concomitant conventional synthetic disease-modifying anti-rheumatic drugs (csDMARD) use.
Methods SPIRIT-H2H is a Wk52 multicentre, open-label, blinded-assessor study comparing IXE and ADA in bionaïve patients with PsA. Patients were randomised 1:1 to IXE or ADA with stratification by concomitant csDMARD use and presence of moderate-to-severe plaque psoriasis. Prespecified end points at Wk24 and Wk52 included musculoskeletal, psoriasis, quality-of life outcomes, subgroup analyses and safety.
Results A significantly higher proportion of patients treated with IXE versus ADA simultaneously achieved ACR50 and PASI100 (39% vs 26%, p<0.001), PASI100 (64% vs 41%, p<0.001) at Wk52. Efficacy of IXE and ADA was similar at Wk52 for ACR50 (49.8% vs 49.8%, p=0.924), treat-to-target outcomes, enthesitis and dactylitis resolution. Responses to IXE were consistent irrespective of concomitant csDMARD use. Significantly more patients on IXE monotherapy versus ADA monotherapy had simultaneous ACR50 and PASI100 (38% vs 19%, p=0.007), and PASI100 responses (66% vs 35%, p<0.001) at Wk52. There were no new safety findings for IXE or ADA.
Conclusions IXE provided significantly greater simultaneous joint and skin improvement than ADA through Wk52 in bionaïve patients with PsA. IXE showed better efficacy on psoriasis and performed at least as well as ADA on musculoskeletal manifestations. IXE efficacy was consistent irrespective of concomitant csDMARD use.
Trial registration number NCT03151551.
- inflammation
- arthritis, psoriatic
- adalimumab
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Prior to disclosure of the primary results of SPIRIT head-to-head (H2H) trial comparing efficacy and safety of ixekizumab with adalimumab in bionaïve patients, direct comparative data were lacking in psoriatic arthritis (PsA).
Ixekizumab was shown to be superior to adalimumab for simultaneous achievement of American College of Rheumatology (ACR)50 and Psoriasis Area and Severity Index (PASI)100 responses along with non-inferiority for ACR50 and superiority for PASI100 achievement at week 24.
What does this study add?
This work presents final SPIRIT-H2H data through 52 weeks: significantly higher simultaneous ACR50 and PASI100 responses as well as PASI75/90/100 responses on ixekizumab than adalimumab were maintained.
At week 52, treatment with ixekizumab was at least as efficacious as adalimumab based on the ACR20/50/70 responses, PsA-specific composite disease activity measures, enthesitis and dactylitis as well as physical function outcomes, with faster effects seen until week 24 for most outcomes.
Ixekizumab efficacy was consistent throughout 52 weeks either as monotherapy or in combination with csDMARDs.
There were no new safety findings for either ixekizumab or adalimumab.
Key messages
How might this impact on clinical practice or future developments?
SPIRIT-H2H study comparing ixekizumab versus adalimumab is the first fully disclosed direct head-to-head study in PsA; its findings over 52 weeks will inform future treatment recommendations and may impact selection of therapy in bionaïve patients with active PsA.
Introduction
Psoriatic arthritis (PsA) is a chronic, progressive, immune-mediated, inflammatory disease affecting the musculoskeletal system, skin and nails.1 Occurring in 0.04%–1% of the population, the disease may result in deformities, impaired physical function, decreased quality of life and increased mortality.2 3 Initial treatment traditionally involves non-steroidal anti-inflammatory drugs, glucocorticoids and conventional synthetic disease-modifying anti-rheumatic drugs (csDMARDs).4–6 When csDMARDs are ineffective, biological (b)-DMARDs such as tumour necrosis factor (TNF)-inhibitors, anti-interleukin (IL)-12/23, anti-IL-17A inhibitors or targeted synthetic DMARD are recommended, with TNF-inhibitors being frequently the first-line bDMARD therapy.3 7–10 bDMARDs can be used as monotherapy or in combination with csDMARDs, most commonly methotrexate (MTX). Ixekizumab (IXE) is an immunoglobulin G4 monoclonal antibody that selectively inhibits IL-17A and its efficacy across multiple PsA domains was previously established.11 12 Despite the approval of numerous bDMARDs for the treatment of PsA, no direct comparisons are available to enable evidence-based treatment decisions.
SPIRIT head-to-head (H2H) is a 52-week (Wk) trial evaluating efficacy and safety of IXE and ADA, a TNF-inhibitor, in patients with active PsA naïve to bDMARDs. We reported previously the results of the primary and key secondary end points at Wk24, which were all met, demonstrating superiority of IXE versus ADA for the simultaneous achievement of American College of Rheumatology (ACR)50 and Psoriasis Area and Severity Index (PASI)100 responses, along with superiority for PASI100 responses and non-inferiority for ACR50 responses.13
Here, we report the efficacy and safety results of SPIRIT-H2H study through Wk52, including results of the prespecified subgroup analyses based on concomitant csDMARD use and presence or absence of moderate-to-severe psoriasis.
Methods
Participants
Participants had to be ≥18 years of age, with a confirmed diagnosis of PsA for ≥6 months and fulfilling the Classification for Psoriatic Arthritis (CASPAR) criteria. Other major inclusion criteria were: previous inadequate response to ≥1 csDMARD, no prior exposure to bDMARDs, ≥3/68 tender joints, ≥3/66 swollen joints, and psoriatic skin lesions affecting a body surface area (BSA) of ≥3%.
Study design
SPIRIT-H2H is a Wk52, phase IIIb/IV, multicentre, randomised, open-label, parallel-group, assessor-blinded study evaluating the efficacy and safety of IXE versus ADA. The detailed study design was published previously.13 Randomisation was stratified by concomitant csDMARD use and presence of moderate-to-severe plaque psoriasis (defined as PASI ≥12 combined with a static Physician Global Assessment ≥3 (of 5) and BSA ≥10%) at baseline (yes/no). Participants received approved label dosing of the assigned treatment based on the presence or absence of moderate-to-severe psoriasis as described in online supplementary information.13 Permitted csDMARDs were MTX (oral or parenteral, 10–25 mg/week), leflunomide (up to 20 mg/day), sulfasalazine (up to 3 g/day) or ciclosporin (up to 5 mg/kg/day) initiated at least 12 weeks prior to baseline and at stable doses as described.13 No change in csDMARD dose was allowed during the first 24 weeks of the study. Study visits took place at screening, baseline (week 0) and postbaseline at weeks 4, 8, 12, 16, 24, 32, 40 and 52.
Supplemental material
Efficacy end points
The primary end point was superiority of IXE to ADA as measured by the proportion of patients who simultaneously achieved ACR50 and PASI100 responses at Wk24. Non-inferiority of IXE compared with ADA for ACR50 response and superiority for PASI100 response at Wk24 were key secondary end points. An ACR50 response was defined as an improvement by ≥50% from baseline in the tender joint count, swollen joint count and ≥3 of the following five measurements: Health Assessment Questionnaire-Disability Index (HAQ-DI), C reactive protein, patient’s assessment of pain Visual Analogue Scale (VAS, 100 mm), patient’s global assessment of disease activity VAS and physician’s global assessment of disease activity VAS. A PASI100 was defined as 100% improvement from baseline PASI score. Nine patients with active psoriasis and BSA ≥3% were assessed as PASI=0 at baseline, a medical inconsistency that was resolved using medical judgement and were considered PASI100 responders if PASI=0 and BSA=0 at postbaseline visits. Multiple analyses to assess the robustness of this approach were conducted.
Prespecified outcomes at Wk52 included the proportion of patients achieving simultaneous ACR50 and PASI100 responses; ACR20/50/70 responses; Minimal Disease Activity (MDA); modified Composite Psoriatic Disease Activity Index (mCPDAI) change from baseline; resolution of enthesitis, as measured by the Spondyloarthritis Research Consortium of Canada Enthesitis Index (SPARCC Enthesitis Index=0) and Leeds Enthesitis Index (LEI=0) among patients with enthesitis at baseline (SPARCC Enthesitis Index >0 or LEI >0 at baseline); resolution of dactylitis, as measured by the Leeds Dactylitis Index-Basic (LDI-B=0) among patients with dactylitis at baseline (LDI-B >0); PASI 75/90/100 responses, resolution of fingernail psoriasis (Nail Psoriasis Area and Severity Index (NAPSI) score=0) and change from baseline in NAPSI score; quality-of-life measures, namely Dermatology Life Quality Index score of 0 or 1 (DLQI (0, 1)), achievement of minimal clinically important difference (MCID) of ≥0.35-point improvement from baseline in HAQ-DI (among patients with ≥0.35 at baseline) and Short Form-36 Physical Component Summary (SF-36 PCS). Post hoc analyses included outcomes based on Disease Activity in PSoriatic Arthritis (DAPSA) and Very Low Disease Activity (VLDA) scores.
Safety
Treatment-emergent adverse events (TEAEs) were defined as an event that first occurred or worsened in severity after the first dose of study treatment and on or prior to the date of the last visit within the treatment time. Adverse events (AEs) of special interest included cytopenias, infections, injection site reactions, allergic reactions, cerebrocardiovascular events, malignancies, depression, inflammatory bowel disease (IBD) and interstitial lung disease. Cardiovascular events and suspected IBD were adjudicated by external clinical events committees.
Statistical analysis
Efficacy
Analyses of efficacy end points were performed at the Wk52 database lock for the intent-to-treat population consisting of all randomised patients according to the treatment to which they were assigned at Wk0.
Categorical variables were assessed using logistic regression models with treatment, concomitant csDMARD use at baseline and presence of moderate-to-severe plaque psoriasis at baseline as factors. Patients were considered non-responders if they did not meet the clinical response criteria or had missing clinical response data at a particular time point of analysis. Continuous efficacy and health outcome variables were analysed using mixed effects model of repeated measures analysis. The models included treatment group, concomitant csDMARD use at baseline, presence of moderate-to-severe plaque psoriasis at baseline, visit-as-fixed factors, baseline value as covariate and baseline-by-visit and treatment-by-visit interaction terms. In addition, logistic regression models were used to test treatment-by-subgroup interaction, where treatment, subgroup and the interaction of treatment-by-subgroup were included as factors. The treatment-by-subgroup interaction was tested at the significance level of 0.10. Treatment group effects were evaluated within each category of the subgroup using Fisher’s exact test, regardless of whether the interaction was statistically significant. No multiplicity adjustments were conducted for the Wk52 analysis.
Safety
Descriptive statistics were performed on the safety population, defined as all randomised patients who received ≥1 dose of the study treatment.
Results
Participants
From the 684 screened patients, 566 were randomised equally between IXE and ADA treatments. Overall, 246 (87%) patients treated with IXE and 237 (84%) treated with ADA completed the Wk52 study visit. Concomitant csDMARD use was stable in the majority of the patients over 52 weeks except for three patients stopping MTX treatment after W24 (two ADA, one IXE) and seven patients changing MTX dose after W24 (three ADA, four IXE). Fewer patients treated with IXE than ADA discontinued for AEs, 13 vs 21, respectively (figure 1). Baseline demographics and disease characteristics were balanced between IXE and ADA groups (table 1).
Baseline demographics and disease characteristics

Participant flow diagram through week 52. bDMARD, biological disease-modifying antirheumatic drug; PsA, psoriatic arthritis.
Efficacy
The Wk52 efficacy results are summarised in table 2. A significantly higher proportion of patients treated with IXE achieved simultaneous ACR50 and PASI100 versus ADA (39.2% vs 26.1%; p<0.001). Significant differences were observed as early as Wk8 and maintained throughout the study (figure 2A). IXE and ADA treatment resulted in similar response rates for ACR50 (49.8% vs 49.8%) (figure 2B), ACR20 (69.6% vs 68.9%) and ACR70 (35.3% vs 34.3%) at Wk52 (figure 3A,B).
Efficacy and health outcomes at Wk52

Key clinical response rates through week 52 (non-responder imputation). (A) Percentage of patients achieving simultaneous American College of Rheumatology (ACR)50 and Psoriasis Area and Severity Index (PASI)100 response. (B) Percentage of patients achieving ACR50 response. (C) Percentage of patients achieving PASI100 response. Ixekizumab (IXE) vs adalimumab (ADA): *p<0.05, †p<0.01, ‡p<0.001.

Additional efficacy outcomes—American College of Rheumatology (ACR) and Psoriasis Area and Severity Index (PASI). Percentage of patients achieving (A) ACR20, (B) ACR70, (C) PASI75 and (D) PASI90, through 52 weeks. Ixekizumab (IXE) vs adalimumab (ADA): †p<0.01, ‡p<0.001.
The proportion of patients achieving MDA, VLDA, DAPSA remission and DAPSA low disease activity or remission was not significantly different between IXE and ADA at Wk52. However, faster onset of response to IXE than ADA with significant differences at Wk24 was observed for MDA (47.7% vs 35.3%; p=0.003) (online supplementary figure 1A), VLDA (17.3% vs 10.2%; p=0.015) and DAPSA remission (26.5% vs 18.0%; p=0.016) (online supplementary figure 1B). Patients treated with IXE achieved significantly greater improvements from baseline in mCPDAI compared with ADA (online supplementary figure 1C) at Wk52 (−4.35 vs −3.85; p=0.004), with significant differences observed at the first postbaseline assessment at Wk12 through Wk52. Comparable efficacy of IXE versus ADA at Wk52 was shown for enthesitis resolution as measured by SPARCC (56.6% vs 48.5%), LEI (61.6% vs 57.1%) and dactylitis resolution LDI-B (83.3% vs 81.0%), as well as improvement in physical function by achieving MCID in HAQ-DI (66.7% vs 64.6%), respectively (table 2).
Supplemental material
IXE treatment resulted in significantly higher response rates versus ADA at Wk52 for PASI75 (78.4% vs 68.6%; p=0.008) (figure 3C), PASI90 (72.8% vs 54.1%; p≤0.001) (figure 3D) and PASI100 (64.3% vs 41.3%; p≤0.001), beginning as early as Wk4 and persisting through to end of the trial (p≤0.001) (figures 2C and 3C,D). At Wk52, a similar rate of nail psoriasis resolution was observed. However, IXE-treated subjects achieved significantly greater improvement in NAPSI scores from baseline versus ADA (−17.78 vs −15.08; p=0.005, online supplementary figure 1D). Rapid and significantly greater improvements in skin-related quality of life were also observed on IXE (DLQI (0, 1)) as early as Wk4 through Wk52 (online supplementary figure 1E, table 2).
Subgroup analysis
At Wk52, no differential treatment was observed when used in monotherapy or combination with csDMARD on simultaneous ACR50 and PASI100, ACR50 or PASI100 response rates (interaction p values of 0.269, 0.139 and 0.200, respectively). IXE as monotherapy or in combination with csDMARD showed similar response, while ADA response was influenced by csDMARD use. Simultaneous ACR50 and PASI100 achievement: monotherapy, IXE 37.8%, ADA 19.0%, p=0.007; combination therapy, IXE 39.9%, ADA 29.1%, p=0.026. ACR50: monotherapy, IXE 51.1%, ADA 41.7%, p=0.227; combination therapy, IXE 49.2%, ADA 53.3%, p=0.479. PASI100: monotherapy, IXE 65.6%, ADA 34.5%, p≤0.001; combination therapy, IXE 63.7%, ADA 44.2%, p≤0.001 (figure 4).
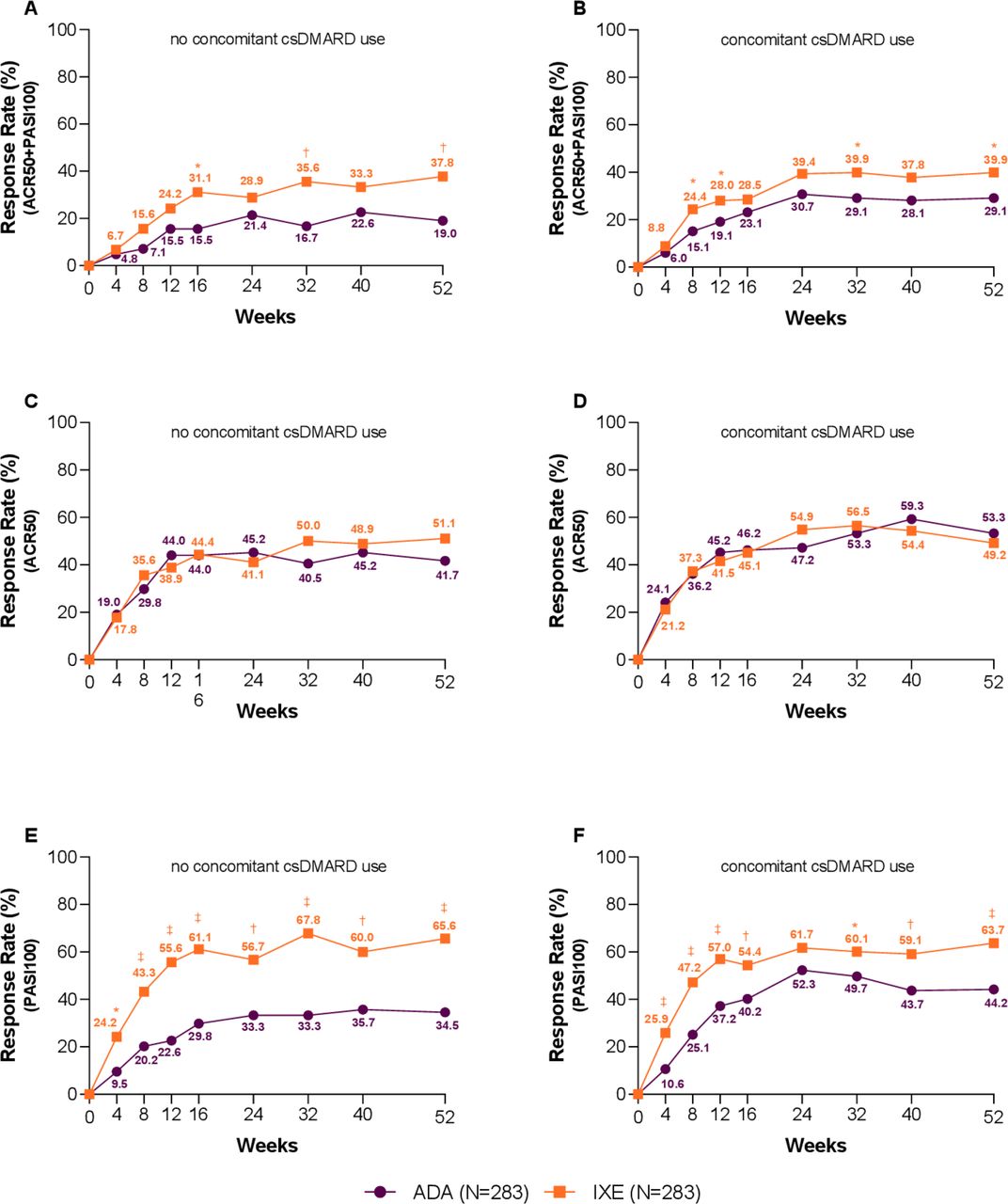
Subgroup analysis based on conventional synthetic disease-modifying anti-rheumatic drug (csDMARD) use—clinical response rates for the key outcomes. Percentage of patients achieving simultaneous American College of Rheumatology (ACR)50 and Psoriasis Area and Severity Index (PASI)100 responses (A) without concomitant csDMARD use and (B) with concomitant csDMARD use (treatment-by-subgroup interaction p=0.269). Percentage of patients achieving ACR50 (C) without concomitant csDMARD use and D) with concomitant csDMARD use (treatment-by-subgroup interaction p=0.139). Percentage of patients achieving PASI100 (E) without concomitant csDMARD use and (F) with concomitant csDMARD use (treatment-by-subgroup interaction p=0.200). Ixekizumab (IXE) vs adalimumab (ADA): *p<0.05, †p<0.01, ‡p<0.001.
Similar observations were made when looking at the difference between IXE and ADA across severity of psoriasis at baseline, where the different dosing of IXE and ADA administered in patients with PsA with/without moderate-to-severe psoriasis did not influence IXE efficacy as measured by simultaneous ACR50 and PASI100 response rate (interaction p=0.277): with moderate-to-severe psoriasis, IXE 38.8%, ADA 17.6%, p=0.026; without moderate-to-severe psoriasis, IXE 39.3%, ADA 28.1%, p=0.014. ACR50 (interaction p=0.378): with moderate-to-severe psoriasis, IXE 55.1%, ADA 62.7%, p=0.542; without moderate-to-severe psoriasis, IXE 48.7%, ADA: 46.8%, p=0.711. PASI100 (interaction p=0.199): with moderate-to-severe psoriasis, IXE 59.2%, ADA 25.5%, p≤0.001; without moderate-to-severe psoriasis, IXE 65.4%, ADA 45.0%, p≤0.001; at Wk52 (figure 5).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Subgroup analysis based on presence/absence of moderate-to-severe psoriasis—clinical response rates for the key outcomes. Percentage of patients achieving simultaneous American College of Rheumatology (ACR)50 and Psoriasis Area and Severity Index (PASI)100 (A) with moderate-to-severe psoriasis and (B) without moderate-to-severe psoriasis (treatment-by-subgroup interaction p=0.277). (C) Percentage of patients with moderate-to-severe psoriasis achieving ACR50 and (D) patients without moderate-to-severe psoriasis achieving ACR50 (treatment-by-subgroup interaction p=0.378). (E) Percentage of patients with moderate-to-severe psoriasis achieving PASI100 and (F) patients without moderate-to-severe psoriasis achieving PASI100 (treatment-by-subgroup interaction p=0.199). Ixekizumab (IXE) vs adalimumab (ADA): *p<0.05, †p<0.01, ‡p<0.001.
Safety
There were numerically more TEAEs in the IXE-treated population compared with ADA (73.9% vs 68.6%, p=0.194) mostly classified as mild or moderate (table 3). There were fewer severe TEAEs occurring in the IXE-treated patients versus ADA (3.2% vs 7.1%), serious AEs (4.2% vs 12.4%) and discontinuations due to AEs (4.2% vs 7.4%). No deaths occurred during the study. Serious infections were numerically lower in IXE treatment compared with ADA (1.8% vs 2.8%), while the number of Candida infections was higher in the IXE-treated group (2.5% vs 1.1%). There was one case each of legionella pneumonia and lymph node tuberculosis reported in the ADA-treated group.
Safety outcomes
Injection site reactions were more frequent in the IXE-treated group versus ADA (10.6% vs 3.5%) while the number of discontinuations due to injections site reaction was lower in IXE versus ADA (0.7% vs 1.1%). The number of hypersensitivity reactions and cerebrocardiovascular events were similar across both groups. Four cases of malignancy were reported in the ADA-treated population (2 basal cell carcinoma, rectal carcinoma and gastrointestinal stromal tumour). No malignancies occurred in the IXE-treated group. One patient discontinued due to rectal carcinoma in the ADA-treated group. Fewer events of cytopenia were observed in the IXE versus ADA-treated group (3.2% vs 4.2%).
Two cases of IBD reported in the IXE-treated group during the period of Wks0–24 were adjudicated. One case of Crohn’s met the EPIdémiologie des Maladies de l’Appareil Digestif (EPIMAD) criteria of confirmed IBD and one case of ulcerative colitis did not meet EPIMAD criteria of confirmed IBD as it was adjudicated as possible.13 No new case was reported during Wks 24–52 period.
Discussion
Although several bDMARDs with different mechanisms of action are approved for use in PsA, true head-to-head trials against an active agent and not versus placebo are still lacking. ADA has previously been included as an active reference arm in SPIRIT-P1 (NCT01695239; IXE vs placebo) and OPAL (NCT01877668; tofacitinib vs placebo) studies; however, these trials were not statistically powered for direct comparisons.11 14 SPIRIT-H2H is the first completed PsA trial directly comparing two bDMARDs, IXE and ADA, in patients with active PsA and an inadequate response to csDMARD/s. This study met the primary end point at Wk24 by demonstrating the superiority of IXE over ADA for the simultaneous achievement of ACR50 and PASI100. The key secondary end points of non-inferiority of IXE for ACR50 and superiority for PASI100 at Wk24 were also met.13 The present work reports the Wk52 results of SPIRIT-H2H, including results of the prespecified subgroup analyses with respect to the concomitant csDMARD use or presence/absence of moderate-to-severe psoriasis. Significantly higher proportions of patients treated with IXE versus ADA simultaneously achieving ACR50 and PASI100 responses were sustained through Wk52. Importantly, response rates for IXE were consistent irrespective of concomitant csDMARD use, while numerically higher response rates for ADA were seen when used in combination with csDMARDs than in monotherapy.
Similar to SPIRIT-H2H, a study comparing secukinumab versus ADA (NCT02745080, EXCEED 1) has enrolled bDMARD-naïve patients with inadequate response to csDMARDs. However, key differences between the two studies include primary outcome (ACR20 in EXCEED vs ACR50+PASI100 in SPIRIT-H2H), blinding (double-blind in EXCEED 1 vs open-label design with blinded assessments in SPIRIT-H2H) and concomitant csDMARD use, which was not allowed in EXCEED 1. The results of EXCEED 1 were only presented in an abstract form thus far.15
Due to disease heterogeneity in PsA, the requirement of assessing multiple PsA domains to identify appropriate treatments for individual patients is important. Therefore, we used simultaneous achievement of a relatively rigorous end point for articular disease (ACR50) and a very stringent end point for the skin disease (PASI100) at Wk24 as the primary end point for this study. IXE treatment demonstrated significantly higher rates for the simultaneous achievement of ACR50 and PASI100 as early as Wk8 and throughout Wk52, highlighting the maintenance of comprehensive disease control throughout the study. A significantly higher PASI100 response rate and change from baseline in NAPSI score was shown in the IXE group as early as the first postbaseline assessment (Wk4 for PASI100 and Wk12 for NAPSI) and through Wk52. IXE and ADA treatment resulted in similar responses in other PsA domains at Wk52: ACR20/50/70; MDA, VLDA, DAPSA remission; enthesitis and dactylitis resolution. At Wk24, significantly higher proportions of patients on IXE achieved resolution of enthesitis (by SPARCC), MDA, VLDA and DAPSA remission, indicating a faster onset of action of IXE versus ADA on the musculoskeletal symptoms of PsA, not only on psoriasis as evidenced by PASI responses and NAPSI resolution. Significantly larger improvement from baseline in mCPDAI was observed in the IXE treatment group compared with ADA, starting at the first postbaseline assessment at Wk12 and persisting to Wk52.
The beneficial effect of the concomitant use of csDMARDs in patients with PsA treated with bDMARDs is still debated. Therefore, concomitant use of csDMARDs was allowed during the study, with the requirement for stable doses 8 weeks prior to baseline and until Wk24 (primary end point). Randomisation was also stratified according to concomitant csDMARD use to enable exploring the response-modifying effect of csDMARD on IXE and ADA treatment. Interestingly, concomitant csDMARD use had a response-modifying effect in the ADA group, but not in the IXE group. This was manifested by numerically higher proportions of patients achieving simultaneous ACR50 and PASI100 as well as PASI100 responses on ADA with concomitant csDMARD use compared with monotherapy in contrast to consistent response rates in the IXE group irrespective of concomitant csDMARD use; these observations can inform decision making when considering concomitant csDMARD to IXE or ADA. Interestingly, the differences in the dosing regimen of IXE and ADA with respect to the presence or absence of moderate-to-severe psoriasis had no effect on the response to either IXE or ADA.
Overall, the safety profiles of both drugs were consistent with previous clinical trials and the regulatory labels. However, a number of numerical differences between the treatment groups were identified. Although IXE treatment resulted in more TEAEs, the majority of these were classified as mild and moderate in severity, while more severe TEAEs and serious AEs were observed in the ADA group. The number of serious infections and proportion of patients who discontinued the study due to AEs was also numerically higher with ADA treatment. The injection site reactions were higher in the IXE group, while the number of patients who discontinued due to in injection site reaction was similar. One case of Crohn’s disease was adjudicated and confirmed in the IXE-treated group.
The limitations of the study include the open-label design, which may have contributed to outcome assessment bias, although efforts were made to minimise this by using blinded assessors for key outcomes. On the other hand, open-label design better represents real-world clinical setting where patients are aware of the treatment assignment. Antidrug antibodies to IXE or ADA were not measured, which would have provided insight into the immunogenicity and possible explanation for the differing impact of concomitant csDMARDs.
In summary, IXE treatment resulted in significantly greater simultaneous ACR50 and PASI100 responses versus ADA through 1 year of the study. IXE performed as well as ADA across musculoskeletal PsA domains with a faster onset of action and showed significantly greater efficacy on the skin and nails through Wk52. Concomitant csDMARD use had a response-modifying effect in the ADA group, but not the IXE group; responses to IXE were consistent in monotherapy and combination with csDMARDs. The results of this study are informative for the management of patients with PsA in routine clinical practice.
Acknowledgments
The authors thank Adam Clooney, PhD, a medical writer and employee of Eli Lilly and Company, for writing and editorial support.
References
Footnotes
Handling editor David S Pisetsky
Correction notice This article has been corrected since it published Online First. The footnote symbol for Crohn's disease in table 3 has been corrected.
Contributors All authors contributed to critical revision of the manuscript and gave final approval for submission and publication. JS, PN and PM provided advice on additional analysis to be performed and interpretation of the data. HT, HS-K, MO, RC, JG and PG contributed to the interpretation of the data. SLL contributed to study conception, data acquisition and interruption. SP, LL, MH, CS and IdlT contributed to data acquisition and interruption.
Funding This study was funded by Eli Lilly and Company, which contributed to study design, data collection, data analysis, data interpretation, manuscript preparation and publication decisions.
Competing interests JSS received grants to his institution from AbbVie, AstraZeneca, Eli Lilly and Company, Merck Sharpe & Dohme, Pfizer and Roche and provided expert advice for, or had symposia speaking engagements with, AbbVie, Amgen, AstraZeneca, Astro, Bristol-Myers Squibb, Celgene, Celltrion, Chugai, Gilead, ILTOO Pharma, Janssen, Lilly, Merck Sharp & Dohme, Novartis-Sandoz, Pfizer, Roche, Samsung, Sanofi and UCB. PM has received research grants from AbbVie, Amgen, Bristol Myers Squibb, Janssen, Eli Lilly and Company, Novartis, Pfizer, Sun, UCB, has performed consultation for AbbVie, Amgen, Bristol Myers Squibb, Boehringer Ingelheim, Galapagos, Gilead, GlaxoSmithKline, Janssen, Eli Lilly and Company, Novartis, Pfizer, Sun, UCB and a speaker for AbbVie, Amgen, Janssen, Eli Lilly and Company, Novartis, Pfizer, UCBHT has performed consultation for Novartis, Eli Lilly and Company, AbbVie. HS-K has performed consultation for AbbVie, Amgen, Mylan, Sandoz-Hexal, Eli Lilly and Company. IdlT is a full-time employee and shareholder of Eli Lilly and Company. LL is a full-time employee of Eli Lilly and Company. MH is a full-time employee of Eli Lilly and Company. CS is a full-time employee and shareholder of Eli Lilly and Company. MO is an Honoraria of Eli Lilly and Company and Astellas. RC is a member of speakers’ bureau for AbbVie, Amgen, Pfizer, Eli Lilly and Company, MSD, UCB, Gilead, BMS, Sanofi. JG has received fees for participating in Advisory Boards or conferences from AbbVie, Novartis, Pfizer, Amjen, Eli Lilly and Company, MSD, Celgene, Zanofi and Roche. PG has received research grants, consultation fees or speaker honoraria from AbbVie, Amgen, Biogen, BMS, Celgene, Chugai, Janssen, Eil Lilly and Company, Medac, MSD, Nordic Pharma, Novartis, Pfizer, Sanofi and UCB. SL-L is an employee and shareholder of Eli Lilly and Company. SP is a full-time employee of Eli Lilly and Company. PN has received grants for research and clinical trials and honoraria for advice and lectures on behalf Eli Lilly and Company, BMS, UCB, Sanofi, Roche, Novartis, Pfizer, AbbVie, MSD, Celgene, Gilead, Janssen.
Patient and public involvement Patients and/or the public were not involved in the design, conduct, reporting or dissemination plans of this research.
Patient consent for publication Not required.
Ethics approval SPIRIT-H2H was conducted in accordance with the ethical principles of the Declaration of Helsinki. All patients provided written informed consent, and the study protocol was approved by the ethical review board prior to the start of study-related procedures.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data may be obtained from a third party and are not publicly available. Eli Lilly and Company provides access to relevant anonymised patient-level data from studies on approved medicines and indications as defined by the sponsor-specific information at www.clinicalstudydatarequest.com. Additional study-related documents will be made available, including the study protocol, statistical analysis plan, clinical study report and an annotated case report form. These materials will be available beginning 6 months after the publication is accepted, given approval of the indication in the USA and EU. These materials will be provided to achieve the aims in the provided proposal.