Article Text

Abstract
Objectives Diverse evidence including clinical, genetic and microbiome studies support a major role of the gut microbiome in the common immune-mediated arthropathy, ankylosing spondylitis (AS). We set out to (1) further define the key microbial characteristics driving disease, and (2) examine the effects of tumour necrosis factor-inhibitor (TNFi) therapy upon the microbiome.
Methods The stools from a case–control cohort of 250 Han-Chinese subjects underwent shotgun metagenomic sequencing. All subjects were genotyped using the Illumina CoreExome SNP microarray.
Results Previous reports of gut dysbiosis in AS were reconfirmed and several notable bacterial species and functional categories were differentially abundant. TNFi therapy was correlated with a restoration the perturbed microbiome observed in untreated AS cases to that of healthy controls, including several important bacterial species that have been previously associated with AS and other related diseases. Enrichment of bacterial peptides homologous to HLA-B27-presented epitopes was observed in the stools of patients with AS, suggesting that either HLA-B27 fails to clear these or that they are involved in driving HLA-B27-associated immune reactions. TNFi therapy largely restored the perturbed microbiome observed in untreated AS cases to that of healthy controls, including several important bacterial species that have been previously associated with AS and other related diseases. TNFi therapy of patients with AS was also associated with a reduction of potentially arthritogenic bacterial peptides, relative to untreated patients.
Conclusion These findings emphasise the key role that the gut microbiome plays in driving the pathogenesis of AS and highlight potential therapeutic and/or preventative targets.
- Ankylosing Spondylitis
- Anti-TNF
- Spondyloarthritis
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Clinical, genetic and microbiome evidence supports the contention that ankylosing spondylitis (AS) is influenced by interactions between the gut microbiome and the host immune system.
The majority of microbiome studies to date have been conducted via sequencing of the 16S ribosomal marker gene, limiting the scope and accuracy of downstream analytics.
What does this study add?
This study contributes three major findings in regards to the role of the gut microbiome in AS: (1) the AS gut microbiome is shaped such that cases carry a higher load of peptides known to be presented by HLA-B27. (2) Changes in prevalence of clinically relevant bacterial species and metabolic pathways that are known to be conducive for proinflammatory responses were noted, and (3) tumour necrosis factor-inhibitor (TNFi) therapy was correlated with restoration of the perturbed microbiome, highlighting a potential mechanism of action for these drugs.
How might this impact on clinical practice or future developments?
Therapeutic and/or preventative modulation of the gut microbiome remains an attractive strategy. To achieve this, precise identification of the microbial characteristics potentially driving disease is required.
Introduction
Ankylosing spondylitis (AS) has been shown in both twin and unrelated case/control studies to be highly heritable (twins >90% heritability).1 2 There is substantial evidence suggesting that the interaction between host genetics and gut microbiome is a key driver of the pathogenesis of AS. The high disease heritability indicates that the environmental factors involved in the disease are likely to be ubiquitous. Reactive arthritis is a spondyloarthritis (SpA) sharing many clinical and genetic features with AS and is known to be caused by bacterial infections of the gut or urinary tract; a subset of patients with reactive arthritis go on to develop AS. About 60% patients with AS suffered from subclinical bowel inflammation and 10% of them can be diagnosed as inflammatory bowel disease (IBD).3 There is considerable overlap in the overall heritability of AS and IBD,4 the two diseases are often cofamilial5 and many shared genetic associations have been identified.6 A bioinformatic study showed that AS susceptibility genes specifically enriched in gut cells are also enriched in ‘response to bacterium’ GO term pathway and that AS-associated genetic loci are found disproportionately to lie within epigenetic marks of gene activity in gut tissue and cells.7 Germ-free HLA-B27-transgenic rats and SKG mice are disease-free.8 9 Studies using sequencing-based bacterial profiling of terminal ileal biopsies showed that patients with AS have a distinct microbiome,10 a finding that has subsequently been reproduced studying stool samples in patients with AS and patients with SpA, a broader clinical classification.11 Recently, HLA-B27-positive healthy individuals were shown have a significantly different microbiome, indicating that the gut microbiome may in fact be a driver of AS, as opposed to being secondary to it.12 There has also been suggestive evidence reported that the gut microbiome is associated with differences in AS disease activity.13 In addition, one study compared stool samples of patients with SpA before and 3 months after the tumor necrosis factor-inhibitor (TNFi) treatment onset.14 Although modest changes were found in microbiome alpha-diversity measures after TNFi treatment, no changes in specific bacterial taxa were observed. In summation, the above evidence supports the contention that AS status is influenced by interactions between the gut microbiome and the host immune system.
To date, the mechanisms involved in the interaction between the host immune system and intestinal microbes remain unclear. One hypothesis suggests that HLA-B27 presents specific peptides to CD8+T cells, leading to pathogenic adaptive immune responses (the ‘arthritogenic peptide theory’). The gut microbiota produces a huge variety and number of peptides, and as such, microbial peptides intrinsic to dysbiosis may activate CD8+T cells. In that context, Purcell and colleagues identified 7500 such peptides that bind the eight most common HLA-B27 subtypes.15 Here, we present our findings from a shotgun metagenomics sequencing study undertaken with stool samples collected from 250 Chinese individuals to investigate evidence of dysbiosis in AS, the effect of TNFi treatment on the gut microbiota and to investigate evidence of immunity to HLA-B27-restricted microbial peptides in AS cases.
Materials and methods
Subject recruitment
A total of 127 unrelated Han Chinese AS cases meeting the 1984 modified New York criteria for AS16 were recruited from the Department of Rheumatology and Immunology of Shanghai Changzheng Hospital (Shanghai, China). A total of 123 healthy controls (blood donors on no prescription medications) were recruited from Shanghai. All human studies have been approved by the Research Ethical Committee of Second Military Medical University, and all patients and controls gave informed written consent for their participation in the studies. Clinical and dietary information was collected described in the online supplementary methods.
Supplemental material
Shotgun metagenome sequencing
DNA extracted from faecal samples were extracted and sequenced using an Illumina HiSeq sequencer (Illumina, San Diego, California, USA) with the paired-end 150 bp sequencing model based on >5G raw data output per sample (see the online supplementary material for more detail).
Manual inspection and QC of sequencing reads were conducted using FastQC v10.1.17 Paired-end reads were joined using PEAR v0.9.1018 and adapters were trimmed using Trimmomatic v0.36.19 Contaminant sequences, such as those mapping to human or PhiX genomes, were filtered using Bowtie2 v2.3.420 and the remaining reads were counted and subsampled to an equal sequencing depth of 3 520 000 sequencing reads per sample using SeqTK v1.0.21 MetaPhlAn2 v2.6.022 was used for taxonomic classification, PanPhlAn v1.2.223 was used for strain-level profiling using precomputed pan-genome references where possible, and HUMAnN2 v0.11.124 was used for functional mapping to KEGG Orthogroups (KO) and MetaCyc pathways and using a UniRef90 database.
For the prediction of bacterial peptides homologous to previously reported HLA-B27-presented epitopes, all available bacterial-derived sequencing reads were BLASTXed against a local, BLAST-formatted,25 version of the immune epitope database (IEDB) v3.0 (downloaded August 2016).26 27 BLAST best-hits with an E-value <0.1, as well as their corresponding IEDB annotations, were collected and counted. The analysed version of IEDB contained 7500 peptides which were previously identified as HLA-B27-presented via proteomics.15 The abundance of each HLA-B27-presented peptide in cases vs controls was evaluated by Wilcoxon rank-sum testing (false discovery rate (FDR) corrected) and the presence/absence of the peptides in cases vs controls was evaluated via the Fisher exact test (FDR corrected), with Q<0.05 considered significant.
Further microbiome analyses were performed using R as outlined in the online supplementary methods.
T-cell responses to microbiome-derived peptides
Individual peptides, previously shown to be presented by HLA-B27 and found in AS case stool microbiomes, were used to pulse peripheral blood mononuclear cells (PBMCs) from either HLA-B27-positive or HLA-B27-negative donors. Pulsed PBMCs were cocultured with autologous PBMC in T-RF10+interleukin-2 (10 U/mL). On day 20 of culture, cells were restimulated with peptide-pulsed irradiated autologous lymphoblastoid cells, and CD4 T cells depleted by magnetic sorting. Ten to 14 days later, interferon-gamma (IFN-γ) production by CD8 T cells was determined by intracellular cytokine staining. Two runs were performed, each run involving different peptides, for both the HLA-B27-positive and HLA-B27-negative donors.
Results
Gut dysbiosis in AS
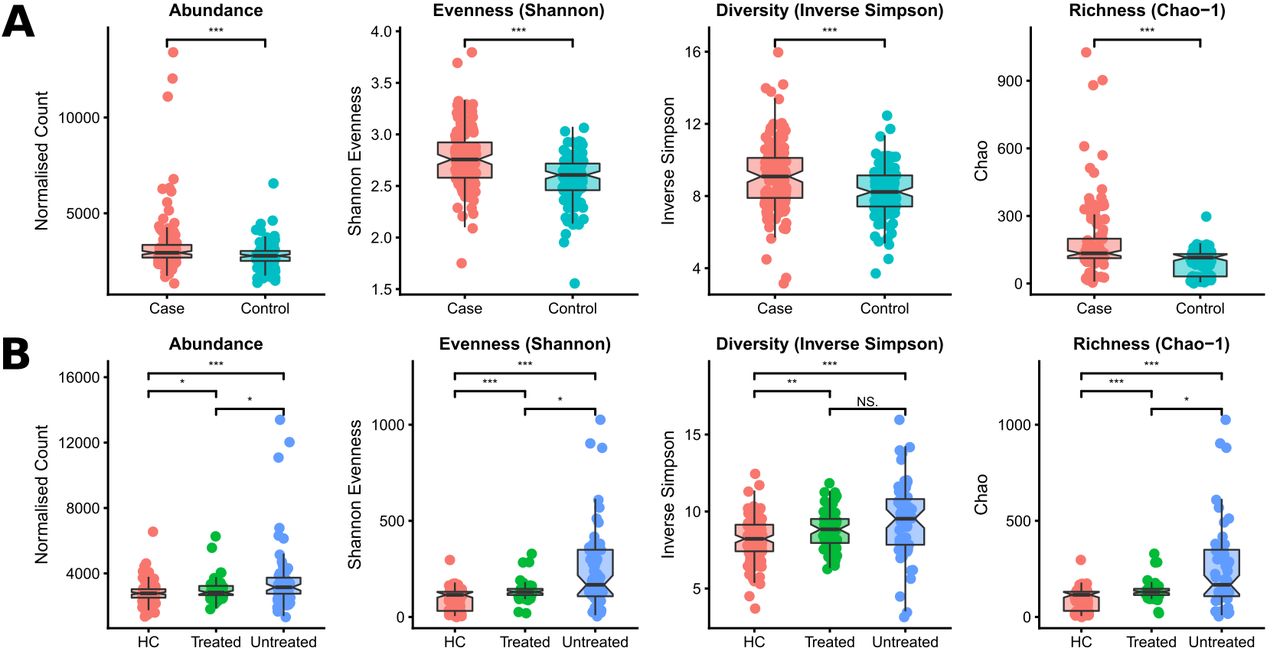
The case and control cohorts were divided into discovery and validation cohorts prior to analysis; the discovery cohort consisted of 97 AS cases and 93 healthy controls with age-matched demographics, and the remaining 60 subjects comprised the validation cohort (30 AS cases and 30 healthy controls) (online supplementary table 1). With the exception of a difference in the mean age in the validation cohorts in which the controls were younger on average than cases, no differences were observed between cases and controls in either the discovery or validation cohorts. PERMANOVA and sPLSDA multivariate analyses revealed significant differentiation between the microbial composition of AS cases and healthy controls for both the discovery (p=0.019) and validation (p=0.0006) cohorts (figure 1A), consistent with previous reports. Receiver-operator curve analysis showed high discrimination between cases and controls using microbiome data alone (area under curve (AUC)=0.87 in combined discovery and validation cohorts) (online supplementary figure 1).
Supplemental material

Taxonomic and functional dysbiosis observed in as cases relative to healthy controls. (A) Alpha and beta diversity analysis. sPLSDA and PERMANOVA revealed community-level differences in taxonomic composition. (B) Commonly differentiated bacterial species from the discovery and validation cohorts. For visualisation purposes, data from the discovery and validation cohorts were combined. (C) Commonly differentiated KEGG Orthogroups from the discovery and validation cohorts. For visualisation purposes, data from the discovery and validation cohorts were combined. Bacterial species and KEGG Orthogroups exhibiting significant results according to multivariate linear modelling and Wilcoxon rank-sum tests are shown. AS, ankylosing spondylitis.
Seven bacterial species were identified to be differentially abundant (p<0.05) (ie, were ‘indicator species’) between AS cases and healthy controls, in both the discovery and validation cohorts (figure 1B). Clostridiales bacterium 1 7 47FAA, Clostridium bolteae and Clostridium hatheway were found to be enriched in AS cases, while Bifidobacterium adolescentis, Coprococcus comes, Lachnospiraceae bacterium 5 1 63FAA and Roseburia inulinivorans were depleted. Several other differentially abundant species of interest were identified in either the discovery or validation cohort, notably Prevotella copri, Dialister invisus and Faecalibacterium prausnitzii. A full list of differentially abundant taxa in either cohort is available in the online supplementary table 2.
Six KO were also found to be differentially abundant (p<0.05) in both cohorts (figure 1C), however, there were no MetaCyc metabolic pathways which were differentially abundant in both cohorts. The commonly differentiated KO were EC 2.6.1.9: histidinol-phosphate transaminase, EC 2.7.4.1: polyphosphate kinase, EC 4.3.3.6: pyridoxal 5'-phosphate synthase, EC 1.15.1.1: superoxide proteinase, EC 3.4.21.53: ATP-dependent serine phosphatase and EC 2.4.2.17: ATP phosphoribosyltransferase. Full lists of the differentially abundant KO and MetaCyc metabolic pathways are available in the online supplementary tables 3 and 4, respectively. All indicator species, except for Lachnospiraceae bacterium 5 1 63FAA, were significantly associated (p<0.05) with the KO, however, the degree of variation explained by these species was typically low with R 2 values ranging from 0.0008 to 0.13 (0.043 on average) (online supplementary table 5).
Strain-level profiling of the dysbiotic microbes identified in figure 1B uncovered no differences in strain composition between AS cases and healthy controls (online supplementary figure 2). This suggests that gut dysbiosis may primarily be a result of differential abundance at the species level and that functional or metabolic differences in the microbiome occur from common genetic elements among the strain population, as evidenced by KO being detectable in the majority of samples in figure 1C.
Effect of TNFi therapy upon the microbiome
To test the effect of TNFi therapy on the gut microbiome, the discovery and validation cohorts were combined to enable sufficient sample sizes for the following categories: healthy controls (n=123), AS cases treated with TNFi (either etanercept or infliximab, n=67) and AS cases who have not received TNFi treatment (n=60). No statistically significant effect of sulfasalazine treatment was observed (p=0.76, online supplementary figure 3), possibly due to sample size constraints. Multivariate comparison of TNFi untreated and treated cases revealed an effect of TNFi treatment on the overall composition of the microbiome (p=0.022) (figure 2A). Untreated cases were significantly different to healthy controls (p=0.0002), whereas treated cases were not significantly different to healthy controls (p=0.069), highlighting a correlation between treatment and restoration of the perturbed composition of the microbiome.

Effect of TNFi therapy on the microbiome. (A) Alpha and beta diversity analysis. sPLSDA and PERMANOVA revealed community-level differences in taxonomic composition. (B) Bacterial species modulated by the effects of TNFi treatment. Bacterial species exhibiting significant results according to multivariate linear modelling and Wilcoxon rank-sum tests are shown. TNFi, tumour necrosis factor-inhibitor.
To identify the key species modulated by the effects of TNFi therapy, species which were both (a) perturbed in untreated AS cases relative to healthy controls, and (b) differently abundant in treated cases compared with untreated cases, were first identified (figure 2B, supplementary table 6). Six of the eight identified species exhibited significant depletion in untreated AS cases, however, TNFi treatment was correlated with restoration of these species to levels indistinguishable from healthy controls. These species were: P. copri, F. prausnitzii, Bilophila unclassified, Klebsiella pneumoniae, Ruminococcus bromii and Eubacterium biforme. The remaining two species (Clostridium symbiosum and Eggerthella unclassified) were enriched in untreated AS and their abundance was no longer different to healthy controls in treated cases. The findings in relation to P. copri and K. pneumoniae were of particular interest given their previous association with rheumatoid arthritis (RA) and AS, respectively, as was the highly abundant (approximately 20% of total bacterial DNA, on average) F. prausnitzii for its notable depletion in several autoimmune diseases.28 TNFi therapy appeared to partially normalise the dysbiotic bacterial species and KO observed in AS cases relative to healthy controls shown in figure 1B and C, however no statistically significant differences between treated and untreated cases were observed, potentially due to sample size constraints (online supplementary figure 4).
The above approach was also used to identify metabolic pathways modulated by TNFi therapy. Twenty MetaCyc metabolic pathways were identified in total and the perturbed abundance observed in untreated AS cases was restored to healthy control levels in 17 of these. In broad terms, these pathways primarily related to amino acid biosynthesis (notably branched-chain and aromatic amino acid biosynthesis), carbohydrate metabolism (notably starch degradation), nucleotide biosynthesis, metabolite biosynthesis and cell structure. Specific details of the 20 differentially abundant MetaCyc pathways are available in the online supplementary table 7.
Linear regression was used to investigate the association between the modulated species and modulated pathways (online supplementary table 8). Except for PWY-6545, pyrimidine biosynthesis which was not associated with any individual identified species, all the pathways were significantly associated with at least two of the identified species. Similarly, all the species were significantly associated with multiple pathways, however the abundances of Bilophila unclassified and K. pneumonieae were inversely correlated with pathway abundance. An increase in K. pneumonieae was associated with a decrease in the abundance of PWY-6737: starch degradation (p=0.014; R 2=−0.043). The observed decrease in the starch degradation pathway for untreated AS cases is primarily attributed to a depletion of F. prausnitzii (p=2.38×10-24; R 2=0.31). Faecalibacterium prausnitzii also exhibited strong associations with other metabolic pathways.
Strain-level profiling of the bacterial species outlined in figure 2B also revealed no discernible differences in strain composition between healthy controls, treated cases and untreated cases, indicating the TNFi therapy was associated with changes in the relative abundance of each species, not necessarily the underlying strain composition (online supplementary figure 5).
Bacterial-derived HLA-B27 epitopes in as cases and healthy controls
To investigate the potential effects of HLA-B27 to ‘shape’ the gut microbiome, we assessed the abundance of bacterial peptides homologous to HLA-B27 epitopes in AS cases and healthy controls. Translated nucleotide searches were performed against IEBD v3.0, annotated according to a published study15 and counted. Significant enrichment of these peptide sequences was observed in AS cases, with 24 of these enriched in both the discovery and validation cohorts (online supplementary table 9). AS cases not only exhibited enrichment of these peptides but the overall diversity of peptides was also increased, with Shannon, Inverse Simpson and Chao diversity indices revealing significant differences between AS cases and healthy controls (figure 3A).

(A) Enrichment, both in terms of abundance and diversity, of bacterial peptides homologous to HLA-B27 epitopes in as cases relative to healthy controls. (B) Differential abundance and diversity of bacterial peptides homologous to HLA-B27 epitopes in TNFi-treated and TNFi-untreated cases and healthy controls. HC, healthy control; TNFi, tumour necrosis factor-inhibitor.
The overall abundance and diversity of bacterial peptides homologous to HLA-B27-presented epitopes were also significantly different between different TNFi treatment categories (figure 3B). Untreated AS cases exhibited increased abundance and diversity of peptides. For patients who underwent TNFi therapy, a reduction in these peptides was observed relative to untreated cases, however their levels remained marginally higher than healthy controls.
T-cell responses to HLA-B27-presenting bacterial peptides
In a preliminary study, CD8 responses to peptides known to be presented by HLA-B27 and found in the AS case stool microbiomes were assessed in two lines from different patients. CD8 responses as assessed by IFN-γ production were observed in the HLA-B27-positive donor to multiple peptides (figure 4). No such responses were noted in the HLA-B27-negative donor (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
CD8 T-cell responses to peptide stimulation. intracellular interferon-γ responses are reported for synthetic peptides listed in the online supplementary table 9. Data are presented for PBMCs alone with no peptide addition (‘T-cell only’), PBMCs with LCLs added at day 10–14 with no peptide addition (‘APC only’), and with both LCL and peptide addition (other lanes). Open columns refer to findings from an HLA-B*2704/B*4006 as case, hatched columns refer to findings from an HLA-B*1502/B*4001 healthy control. Panels A and B represent separate experiments with different peptides added. APC, antigen presenting cell; IFN, interferon; LCL, lymphoblastoid cell line; PBMC, peripheral bloodmononuclear cell.
Discussion
Gut dysbiosis in AS
This study reconfirmed the occurrence of bacterial gut dysbiosis in AS cases and identified seven bacterial species which were commonly differentiated between cases and controls in both the discovery and validation cohorts (figure 1B). Two of these species, Bifidobacterium adolescentis and Coprococcus comes, have been noted for their depletion in Crohn disease29 and were also observed to be depleted in AS cases in this study. An additional two species previously reported to be associated with AS, P. copri and D. invisus, were found to be differentially abundant in either the discovery or validation cohorts (online supplementary table 2). In the case of P. copri, previous studies have demonstrated enrichment in new-onset RA cases yet depletion in chronic RA cases.30 Consistent with these findings, our study found that P. copri was depleted in AS cases within the non-age-matched cohort, for which the demographics were heavily skewed towards older patients with AS with the long-standing disease (online supplementary table 1). Previous studies in AS have shown increases in Prevotellaceae 10 or specifically with this species.11 As discussed below, P. copri carriage normalised with TNFi treatment. Further studies will be required to determine if P. copri carriage changes with disease duration, as has been reported in RA.
Carriage of Dialister species has been previously associated with disease activity in patients with SpA,13 but the carriage of D. invisus has been reported to be decreased in IBD.31 32 While we found enrichment of D. invisus in AS cases in the discovery cohort, this was not confirmed in the validation cohort, nor has it been reported in other AS studies. Its pathogenic significance is therefore uncertain.
Of particular interest, the notable ‘peace keeping’ microbe F. prausnitzii was also found to be depleted in AS cases in the validation cohort. This bacterium has also been consistently shown to be depleted in IBD32–37 and has been previously shown to be depleted in the disease enthesitis-related arthritis, a paediatric disease-classification which includes children with AS.38 Faecalibacterium prausnitzii is known to produce butyrate and other metabolites and peptides that have diverse anti-inflammatory effects including promoting T-regulatory cell differentiation,39 influences on Th17 lymphocyte activation and promotion of gut mucosal barrier function.37 40 41 As discussed below, TNFi treatment also led to normalisation of F. prausnitzii carriage. These findings suggest that F. prausnitzii plays a key anti-inflammatory role in AS, as it does in IBD.
Using metagenome sequencing to provide a functional characterisation of the microbiome, this study shows that genes encoding pyridoxal 5'-phosphate synthase, an important enzyme for the biosynthesis of vitamin B6, were less abundant among the microbiome of AS cases compared with healthy controls in both cohorts. Vitamin B6 plays a role in the maintenance of vitamin homeostasis in colonocytes.42 43 It has been found to modulate colonic inflammation and several studies that have investigated the role of vitamin B6 for the treatment of inflammation in patients with RA.44–49 Evidence from case-control studies show that patients with RA have low vitamin B6 status compared with healthy controls, however, intervention studies have yielded inconsistent findings, possibly due to the dose of the administered vitamin B6. The reduced potential for the microbiome of patients with AS to produce pyridoxal 5'-phosphate synthase, and thus vitamin B6, may warrant further investigation into intervention strategies to mitigate inflammation in patients with AS.
MetaCyc pathway analysis revealed the depletion of the aromatic and branched-chain amino acid biosynthesis pathways in untreated AS cases, which are responsible for the production of four of the nine essential amino acids in humans (leucine, isoleucine, valine and phenylalanine) (online supplementary table 7). Vitamin B6 is an essential cofactor for branched-chain amino acid transaminase, the last step of the branched-chain amino acid synthesis. Therefore, in the current study, we not only observed depletion of genes encoding the branched-chain amino acid biosynthesis pathway but also for the enzyme which synthesises an important cofactor in the process.
Effect of TNFi therapy upon the microbiome
The previous study of patients with RA before and after synthetic disease-modifying antirheumatic drug treatment revealed moderate differences in the gut microbiota composition, with the perturbed microbial composition being party restored following treatment.50 Similarly, analysis of patients with SpA before and after TNFi therapy also revealed modest differences in microbial composition yet no specific taxon was found to be modulated, likely due to sample size.14 Using a larger sample size, we confirmed that TNFi therapy was correlated with a restoration of the perturbed microbial composition, and additionally identified several notable bacterial species modulated by treatment.
We observed that TNFi therapy restored the depletion of F. prausnitzii in AS cases. Restoration of F. prausnitzii abundance was also correlated with the restored abundance of aromatic and branched-chain amino acid biosynthesis pathways. A recent ulcerative colitis study33 revealed reduced dysbiosis and increased F. prausnitzii abundance in responders compared with non-responders following TNFi therapy. Furthermore, recovery of F. prausnitzii in patients with ulcerative colitis after relapse was associated with maintenance of remission.51 Another study demonstrated that treatment of infliximab completely restored F. prausnitzii concentrations from zero to 1.4×1010 bacteria/mL within a few days.30
Another important microbe, P. copri, was observed to be enriched to levels closely matched to that of healthy controls following TNFi treatment. The abundance of P. copri has previously been shown to be enriched in patients with untreated new-onset RA yet depleted in chronic RA cases, patients with psoriatic arthritis and healthy controls.30 Colonisation of SKG mice with P. copri-dominated microbiota from patients with RA led to an increased number of Th17 cells in the large intestine.52 HLA-DR-presented peptides (T-cell epitopes) from P. copri were recently found to stimulate Th1 responses in 42% of new-onset RA cases, with subgroups of patients with RA demonstrating differential IgG or IgA immune reactivity, providing evidence that P. copri is immune-relevant in RA pathogenesis.53 Additionally, the presence of the HLA-DRB1 risk allele, which influences disease severity, in patients with RA was found to be inversely correlated with P. copri abundance.30 54–56 A recent study of Chinese patients with AS revealed enrichment of P. copri, as well as P. melaninogenica and Prevotella sp. C561.11 Contrasting these results, in the current study, we observed a depletion of P. copri in untreated AS cases, which was restored to the healthy control levels in TNFi-treated patients. These seemingly conflicting reports of P. copri abundance may be explained by the large degree of intraspecific genetic diversity of P. copri strains, with strain variation adding an additional layer of complexity for predicting the function of P. copri in the gut. The Prevotella genus also contains members that may be beneficial, and which do not function as pathobionts,54–56 with observed enrichment in healthy individuals. Taken together, our results which demonstrate modulation of P. copri abundance in TNFi-treated cases are a noteworthy observation, however without a stronger grasp of the strain-level genome variation within this taxon and their prevalence across our cohort attempts to therapeutically modulate and predict the effects of P. copri remain significant challenges.57
Klebsiella pneumoniae has also been suggested to play a significant role in AS pathogenesis,58 although this remains controversial.59 Klebsiella pneumoniae notably produces pullulanase, a starch-debranching enzyme which enables the degradation of starch into simple sugars.60 The apparent arthritogenic effects of dietary starch in AS are based on the concept that the growth of Klebsiella sp. are favoured by these diets and drive AS pathogenesis.61 62 Consequently, low starch diets have been promoted and are frequently followed by patients,62 although there is no published evidence to date as to their efficacy in positively affecting AS disease course. Here, we actually observed depletion of this microbe in untreated cases relative to healthy controls, whereas TNFi-treated cases showed a restoration of this bacterium. Furthermore, our metagenome sequencing data showed an inverse correlation between K. pneumoniae relative abundance and the overall starch degradation metabolic pathway (p=0.014; R 2=−0.043) (online supplementary table 5). This pathway not only includes the pullulanase-mediated starch debranching reaction but also further downstream reactions including the transport and catabolism of maltodextrins. These findings do not support an association between K. pneumoniae and AS pathophysiology, although the role of dietary and/or resistant starches on the gut microbiota and AS warrants further investigation.
Enrichment of bacterial peptides
Pathogenic bacteria have long been hypothesised as an immunological trigger of AS pathogenesis. In the current study, patients with AS not only demonstrated enrichment of bacterial peptides matching HLA-B27 epitopes (online supplementary table 9), but the diversity of these peptides was greater overall (figure 3A). These data provide supporting evidence for the molecular mimicry hypothesis for which bacterial-derived peptides may stimulate AS via cross-activation of autoreactive T cells or B cells, thus leading to autoimmunity. Only limited evidence of CD8 T-cell reactivity to specific antigens has previously been reported in AS63 or the related disease reactive arthritis.64 In our preliminary studies, we showed substantial responses of CD8 T cells to peptides known to be presentable by HLA-B27, and were also found in the stool microbiome of AS cases, in HLA-B27-positive but not HLA-B27-negative donors. Considerable further studies are indicated to investigate this in greater detail including the range of peptides involved, extent of HLA-restriction and differences in T-cell reactivity between AS cases and healthy controls. This hypothesis does not however explain the increase in HLA-B27 epitopes among AS cases, which could be explained by effects of non-HLA genetic factors or AS-associated environmental factors. An alternate hypothesis is that their excess carriage is caused by a deficiency in the ability of HLA-B27 to effectively control their presence, consistent with evidence of increased bacterial migration across the gut mucosa in AS.65 Interestingly, the modulation of the gut microbiome associated with TNFi treatment was also associated with a restoration of the elevated abundance and diversity of peptides observed in untreated cases to levels which were more closely matched to healthy controls (figure 3B). Further research will be required to resolve these alternate hypotheses.
Limitations
Given the known heterogeneity of the gut microbiome in different geographical locations, changes in specific bacterial species may not be replicable in other populations. Furthermore, known technical variability also limits the direct comparison of results between different sequencing methodologies and bioinformatic approaches. A meta-analysis of metagenomics data from diverse populations which have undergone identical data processing procedures will be required in the future to counteract known technical and biological biases. Larger sample sizes may also be required to provide sufficient statistical power, for example, to examine the effect of sulfasalazine on the microbiome. Further work is also required to establish causal relationships between the identified associations. Given the large number of peptides presented by HLA-B27, it is likely that many more peptides other than the ones identified in this study are enriched in the microbiome of patients with AS. Larger sample sizes and continual update of the Immune Epitope Database are likely to expand on our initial observations.
Conclusions
In summary, this study confirms that AS is characterised by gut dysbiosis and identifies key indicator species, several of which are shared with IBD. This dysbiosis is associated with functional differences in the microbiome involving known inflammation-related pathways. We demonstrate that treatment with TNFi, which is highly effective in suppressing the clinical manifestations of AS, is correlated with normalisation of the gut microbiome, and its functional properties, in AS cases. We further demonstrate that the AS gut microbiome is enriched for bacterial peptides that have previously been shown to be presented by HLA-B27 and that this enrichment is also normalised by TNFi treatment. These findings are consistent with disease models in which AS pathogenesis is driven by interactions between a genetically primed host immune system, and the gut microbiome and point to potential therapeutic and/or preventative approaches for the disease.
Acknowledgments
The authors acknowledge and thank the contribution of the subjects who participated in this study.
References
Footnotes
Handling editor Josef S Smolen
JY and PRS contributed equally.
MAB and HX contributed equally.
Correction notice This article has been corrected since it published Online First. The second corresponding author has been added and affiliation 7 has been updated.
Contributors Study design was performed by HX, JY and MAB. Subject recruitment and sample collection was performed by JY, JS, TL, LZ, XW and JZ. Metagenomic analysis was performed by PRS, and bacterial epitope studies by JY, FH and MW. The manuscript was prepared by PRS, MM, MAB and HX.
Funding HX is supported by National Science Foundation of China (Grant 81430031) and China Ministry of Science and technology (973 Program of China 2014CB541800). MAB is funded by a National Health and Medical Research Council Senior Principal Research Fellowship (#1024879).
Competing interests None declared.
Patient consent for publication Not required.
Ethics approval All human studies have been approved by the Research Ethical Committee of Second Military Medical University, and all patients and controls gave informed written consent for their participation in the studies.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement Data are available upon reasonable request.