Article Text

Abstract
Objective The causality and pathogenic mechanism of microbiome composition remain elusive in many diseases, including autoimmune diseases such as rheumatoid arthritis (RA). This study aimed to elucidate gut microbiome’s role in RA pathology by a comprehensive metagenome-wide association study (MWAS).
Methods We conducted MWAS of the RA gut microbiome in the Japanese population (n case=82, n control=42) by using whole-genome shotgun sequencing of high depth (average 13 Gb per sample). Our MWAS consisted of three major bioinformatic analytic pipelines (phylogenetic analysis, functional gene analysis and pathway analysis).
Results Phylogenetic case–control association tests showed high abundance of multiple species belonging to the genus Prevotella (e.g., Prevotella denticola) in the RA case metagenome. The non-linear machine learning method efficiently deconvoluted the case–control phylogenetic discrepancy. Gene functional assessments showed that the abundance of one redox reaction-related gene (R6FCZ7) was significantly decreased in the RA metagenome compared with controls. A variety of biological pathways including those related to metabolism (e.g., fatty acid biosynthesis and glycosaminoglycan degradation) were enriched in the case–control comparison. A population-specific link between the metagenome and host genome was identified by comparing biological pathway enrichment between the RA metagenome and the RA genome-wide association study results. No apparent discrepancy in alpha or beta diversities of metagenome was found between RA cases and controls.
Conclusion Our shotgun sequencing-based MWAS highlights a novel link among the gut microbiome, host genome and pathology of RA, which contributes to our understanding of the microbiome’s role in RA aetiology.
- rheumatoid arthritis
- autoimmune diseases
- gene polymorphism
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key messages
What is already known about this subject?
Rheumatoid arthritis (RA) is one of the diseases for which the microbiome may have an important role in pathology. Gut microbiome has been implied to lead immune abnormality in RA patients such as the activation of immune responses via Th17 cells by Prevotella copri.
What does this study add?
Multiple Prevotella spp. other than P. copri were related to RA etiology in the gut microbiome of the Japanese population.
A redox reaction–related gene (R6FCZ7) was abundant in the gut metagenome of the Japanese patients with RA.
A population-specific biological pathway link between the metagenome and the host genome was identified by comparing the RA metagenome-wide association study (MWAS) and the RA genome-wide association study (GWAS).
Our study indicated a value of metagenome-wide shotgun sequencing rather than classical amplicon sequencing of 16S ribosomal RNA (rRNA) genes of microbiomes.
How might this impact on clinical practice or future developments?
We revealed a novel link between the gut microbiome, host genome and pathology of RA. Our study will be a platform model of the microbiome studies to elucidate etiology of rheumatic diseases.
Introduction
The human microbiome, which refers to the microbial communities inhabiting the human body, has remarkable effects on our immune system and metabolism.1 Different microbiome compositions have been implicated in the pathogenesis of many diseases, such as type 2 diabetes, cardiometabolic disorders, inflammatory bowel diseases and cancer.2–5 Rheumatoid arthritis (RA) is a prevalent autoimmune disorder characterised by synovial inflammation and joint damage.6 In addition to environmental and genetic factors, the microbiome has emerged as a candidate factor responsible for the onset of RA.7 The microbiomes of various sites within the body such as the gut, oral cavity and lungs have been implicated in RA.8–14 Several mechanisms leading to immune abnormality in RA have been reported (e.g, the production of citrullinated peptides by Porphyromonas gingivalis 15 and the activation of immune responses via Th17 cells by Prevotella copri 13). However, the aetiology of RA linked to the microbiome still remains unknown. Additionally, although ethnicity and dietary habits are major factors leading to differences in microbiome compositions, there have been few population-specific microbiome studies focused on RA in non-European or non-Westernised populations.
The spread of microbiome research in academia and industry has contributed to the development of the theoretical and methodological features of bioinformatic analysis methods. While such methods initially focused on amplicon sequencing of 16S ribosomal RNA (rRNA) genes, today it is recommended to perform metagenome-wide association studies (MWAS) based on whole-genome shotgun sequencing of the microbiome. This method can detect the genomic composition of the microbiome at the species level (bacteria, and also archaea, fungi and viruses) and analyse their biological functional features.16 Thus, shotgun sequencing has the potential to indicate new targets for drug therapy as well as faecal transplants and probiotics.17 On the other hand, shotgun sequencing analysis is costly and requires a machine resource that can analyse large data sets as well as complex systematic data processing. As for the gut microbiome in RA, a few studies using shotgun sequencing have been reported,8 11 and findings are not universally consistent.
As previously suggested, shotgun sequencing has the benefit of providing insights into the functional aspects of microbiome genes. RA is one of the representative diseases of which many associated genomic regions have been discovered by genome-wide association studies (GWAS), and a part of its pathogenesis has been elucidated.18 However, the link between the host genetic factor and the microbiome, namely MWAS–GWAS interaction, has not been fully assessed to date.
Unsupervised clustering analyses of taxa or genes have been performed to grasp the overall phylogenetic or functional picture of the metagenome. Machine learning (ML) is one of today’s most rapidly growing technologies. Various clustering approaches that incorporate an ML method have been used, such as principal component analysis (PCA) and the k-means method,19 20 but these classical methods mostly assume linearity of the microbiome data. Recently, ML methods for non-linear dimensionality reduction such as uniform manifold approximation and projection (UMAP)21 have been successfully adopted in diverse analyses.22 Application of such non-linear ML methods should contribute to our advanced understanding of the metagenome.
Here, we report a comprehensive MWAS of the gut microbiome in an RA case–control cohort of the Japanese population. We carried out whole-genome shotgun sequencing of 124 faecal samples (82 individuals with RA and 42 healthy controls). Our MWAS consisted of three major bioinformatic analytic techniques (phylogenetic analysis, functional gene analysis and pathway analysis), which allowed us to comprehensively grasp case–control disparity in the gut metagenome. We newly introduced an unsupervised ML-based clustering approach to depict the RA metagenome landscape. To evaluate the link between the gut metagenome and the human germline genome, we compared the pathway enrichment of the gut microbiome MWAS and that of the host GWAS in RA.
Results
High abundance of multiple species belonging to the genus Prevotella in the RA gut microbiome
We performed whole-genome shotgun sequencing of a total of 124 faecal DNA samples (sequencing group 1: 29 samples from 15 individuals with RA and 14 controls; sequencing group 2: 95 samples from 67 individuals with RA and 28 controls; see online supplementary table 1), which passed stringent quality control (QC) for sequence reads and samples (online supplementary figure 1).
After QC, we assigned sequence reads to taxonomic references and quantified phylogenetic relative abundances separately for each taxonomic level (online supplementary figure 2). We selected a total of 803 clades with an average relative abundance of >1.0×10−5 and detected in more than 20% of the samples in both sequencing groups, including 10 phyla (L2), 23 classes (L3), 34 orders (L4), 72 families (L5), 185 genera (L6) and 479 species (L7). Case–control phylogenetic association tests using a generalised linear regression model identified significant associations of the nine clades (empirically estimated false discovery rate (FDR) q<0.05; figure 1A,B, table 1). Notably, all the associated clades increased in RA samples compared with controls. As illustrated in a phylogenetic tree indicating the case–control association results of multilayered taxonomic levels (figure 1C), clades with more specific taxonomic levels (such as genus (L6) or species (L7)) tended to show greater differences in case–control factors (i.e., effect size). Among the nine clades with significant case–control associations, eight clades belonged to species (L7, the most specific level). Because it was difficult to detect these species level clades using classical 16S rRNA sequence analysis, these results clearly demonstrate the value of metagenome shotgun sequencing in identifying disease-associated taxa. We note that our shotgun sequencing study newly identified that the majority of RA-associated clades belonged to the genus Prevotella (P. denticola, P. marshii, P. disiens, P. corporis and P. amnii). While P. copri has been reported to be abundant in the gut microbiome of patients with RA,13 our results revealed that multiple other species in the genus Prevotella are also related to RA aetiology.

MWAS results of RA case–control phylogenetic association tests. (A) A quantile-quantile plot of the MWAS p values of the clades. The x-axis indicates empirically estimated median −log10 p values. The y-axis indicates observed −log10 p values. The diagonal grey line represents y = x, which corresponds to the null hypothesis. The horizontal red line indicates the empirical Bonferroni-corrected threshold (α=0.05), and the brown line indicates the empirically estimated FDR (q=0.05). Clades with p values less than the Bonferroni thresholds are plotted as red dots. Clades with q<0.05 are plotted as brown dots, and other clades as black dots. (B) A volcano plot. The x-axis indicates effect sizes of generalised linear model. The y-axis, horizontal lines and dot colours are the same as in (A). (C) Phylogenetic tree. Levels L2–L7 are from the inside layer to the outside layer. The size and colour of dots represent relative abundance and effect sizes, respectively. The nine clades with significant case–control associations (q<0.05) are outlined in red. (D) A dimension-reduced plot of the 479 species using UMAP. The eight species with q<0.05 are indicated in red, while the others are shown according to the effect sizes in phylogenetic case–control association tests. (E) Unsupervised clustering results according to the density-based spatial clustering of applications with noise (DBSCAN) algorithm. Seven clusters are illustrated as polygons. Cluster 9, which shows the significant enrichment of species with significant case–control discrepancies, is coloured according to effect sizes (as in figure 1D), while other clusters are shown in black. FDR, false discovery rate; MWAS, metagenome-wide association study; RA, rheumatoid arthritis; UMAP, uniform manifold approximation and projection.
Clades with significant case–control discrepancy
Identification of significant taxa cluster using a non-linear ML method
We performed ML-based deconvolution of the metagenome data to comprehensively assess the case–control discrepancy at the species level (L7). First, we adopted UMAP, a non-linear dimensionality reduction technique, for deconvolution of the complex taxonomic relative abundance data into two-dimensional data (figure 1D). We subsequently performed unsupervised clustering of the results of UMAP with the density-based spatial clustering of applications with noise (DBSCAN) algorithm,23 which classified the species into 14 clusters (online supplementary table 2). Next, we assessed the degree to which each cluster contained species with significant differences in the case–control phylogenetic association tests. One cluster was enriched with species with case–control discrepancies, as shown by hypergeometric tests (p=1.9×10−8, OR=27; figure 1E). Among the eight species which showed q-values <0.05 in the case–control phylogenetic association tests, half belonged to this cluster (n=4). The sum of the species belonging to this cluster also showed a significant difference in the case–control association test (p=0.0057, effect size=0.48). While we parallelly applied classical linear ML methods such as PCA and non-metric multidimensional scaling in the same manner, cluster deconvolution was not clear and no cluster showed a significant case–control discrepancy (online supplementary figure 3). These results indicate that the non-linear ML method could efficiently provide novel knowledge in metagenome analyses.
A redox reaction-related gene of the gut metagenome decreased in RA samples
To evaluate functional aspects of the RA gut metagenome, we performed metagenomic shotgun sequencing according to the following procedures: (1) de novo assembly, (2) prediction of open reading frames (ORFs), (3) clustering and annotation of ORFs, and (4) read mapping to assembled contigs. We selected 179 333 and 211 315 genes annotated by the UniRef9024 and Kyoto Encyclopedia of Genes and Genomes (KEGG)25 databases, respectively. Case–control gene association tests using a generalised linear regression model found significant associations for a total of 12 genes (1 and 11 for UniRef90 and KEGG, respectively; empirically estimated FDR: q<0.05; figure 2, table 2). Of these, nine showed increased abundance in RA samples compared with controls. One gene demonstrated a highly significant association that satisfied the empirical Bonferroni’s correlation (p=2.5×10−7, effect size=−1.19, R6FCZ7). R6FCZ7 is registered as a FeS assembly sulphur utilisation factor system protein in the UniRef database; it has not yet been given an official gene name by nomenclature committees. This kind of protein performs a wide range of bacterial functions, such as electron transfer, redox catalysis and gene regulation.26 Several previous studies have reported that reactive oxygen species play an important role in the pathogenesis of RA,27 and Zhang et al. 8 also reported alteration of the redox environment in the RA gut microbiome. In taxonomic assignment, the source strain of R6FCZ7 was registered as Bacteroides sp . CAG:633. In our metagenome data, the R6FCZ7 sequences were further linked to the taxonomic reference genomes of Bacteroides uniformis, B. rodentium and B. fragilis, as well as Bacteroides sp. This implies that several species belonging to the genus Bacteroides functionally possess this gene. Overall, our results suggest that the redox function of the microbiome, especially the genus Bacteroides, may have an important role in the pathology of RA.

MWAS results of RA case–control gene association tests. (A) A QQ plot (left) and volcano plot (right) of the MWAS p values of genes based on the UniRef90 protein database. (B) A QQ plot (left) and a volcano plot (right) of genes based on the KEGG gene database. In the QQ plots, the x-axis indicates empirically estimated median −log10 p values. In the volcano plot, the x-axis indicates beta of generalised linear model as effect sizes. The y-axis in both plots indicates observed −log10 p values. The diagonal grey line represents y=x, which corresponds to the null hypothesis. The horizontal red line indicates the empirical Bonferroni-corrected threshold (α=0.05), and the brown line indicates the empirically estimated FDR (q=0.05). Genes with p values less than Bonferroni thresholds are plotted as red dots. Clades with q<0.05 are plotted as brown dots, and other clades as black dots. MWAS, KEGG, Kyoto Encyclopedia of Genes and Genomes; MWAS, metagenome-wide association study; QQ, quantile-quantile; RA, rheumatoid arthritis.
Genes with significant case–control discrepancy
Identification of metagenomic biological pathways contributing to the pathophysiology of RA
Using the results of the gene analysis part of our MWAS, we performed gene set enrichment analysis to conduct case–control pathway association tests. We found significant associations for 19 Gene Ontology (GO) terms that satisfied the empirical Bonferroni’s correlation (figure 3A, table 3). One of the significant GO terms was metal ion binding (p=2.0×10−5, GO:0046872), which implies that the interaction between reactive oxygen species and metal ions is associated with the pathology of RA, which is similar to the result of the gene association tests. The eight KEGG pathways showed significant associations (q<0.05; figure 3B). A part of the substances related to these pathways are involved in the pathophysiology of RA as follows: (1) Fatty acids have been found to be related to inflammation and several free fatty acids in serum are reported to be upregulated in patients with RA.28 (2) Terpenoids suppress nuclear factor-kB signalling, the major regulator in the pathogenesis of inflammatory diseases.29 They are the main component of Tripterygium wilfordii, a plant used in traditional Chinese medicine which has been shown to be non-inferior to methotrexate in the treatment of RA.30 (3) Adipocytokines have been reported to contribute to the proinflammatory state of RA, increasing in the synovial fluid of patients with RA.31 (4) Glycosaminoglycans (GAGs) are major components of joint cartilage and other soft connective tissues. Modification of GAG metabolism may play a crucial role in RA pathogenesis.32 These results suggest that the aforementioned substances in the gut could influence the pathology of RA. The system diagram of the KEGG pathways with the results of the case–control pathway association tests clearly illustrates that a variety of pathways, including those related to metabolism, showed significant case–control differences (figure 3C).

MWAS results of RA case–control pathway association tests. (A) QQ plot of the MWAS p values of pathways based on GO terms. (B) A QQ plot of the MWAS p values of pathways based on GO terms. KEGG pathways. (C) System diagram of KEGG pathways. The three levels are defined as A, B and C, and described from the inside layer out. The size and colour of dots represent set sizes and p values, respectively. The eight pathways with significant enrichment (q<0.05) are outlined in red. (D) Comparison of p values of KEGG pathways between the RA MWAS and GWAS data. The x-axis indicates the p values of the GWAS data (left, East Asians; right, Europeans). The y-axis indicates the p values of the MWAS of Japanese. The horizontal and vertical black lines indicate p value of 0.05. The overlap of the pathway enrichment was evaluated by classifying the pathways based on the significance threshold of p<0.05 or p≥0.05 and using Fisher’s exact test. FDR, false discovery rate; GO, Gene Ontology; GWAS, genome-wide association study; KEGG, Kyoto Encyclopedia of Genes and Genomes; MAPK, mitogen-activated protein kinase; MWAS, metagenome-wide association study; QQ, quantile-quantile; RA, rheumatoid arthritis.
Pathways with significant case–control discrepancy
Population-specific shared biological pathways between the metagenome and host genome in RA
We assessed whether biological pathways of the gut metagenome and the host germline genome were shared in RA. In addition to our KEGG pathway enrichment of the RA gut metagenome of the Japanese population, we estimated pathway enrichment in the host genome of RA samples, using previously conducted RA GWAS data in East Asian and European populations (n=22 515 and n=58 284, respectively).18 We note that the majority of subjects in the East Asian RA GWAS were of Japanese ancestry (93.1%). We compared the p values of the KEGG pathways shared between the RA MWAS data (Japanese) and the RA GWAS data (separately for East Asians and Europeans; figure 3D). Several pathways showed significant enrichment between the metagenomes and host genomes (p<0.05 in both; e.g., FoxO signalling pathway, Th17 cell differentiation and interleukin-17 signalling pathway). We observed a significant correlation between pathway p values in East Asians (p Fisher=0.045), but not in Europeans (p Fisher=0.41). Considering that MWAS–GWAS pathway correlation was specifically observed when the metagenome and the host genome data in similar populations were compared, there should be a population-specific link between the germline genome and metagenome in RA pathology.
No apparent discrepancy in metagenome diversity between RA cases and controls
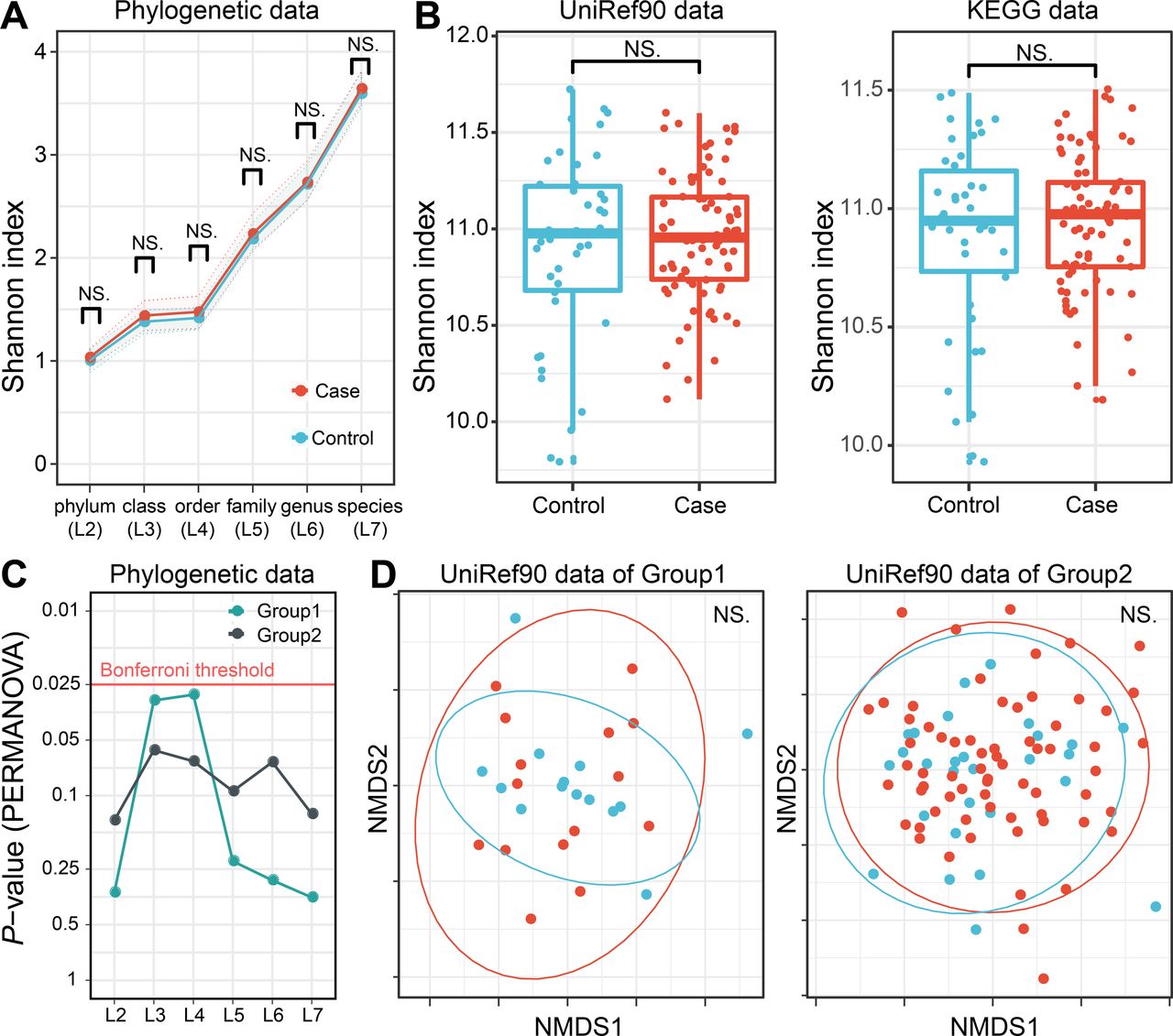
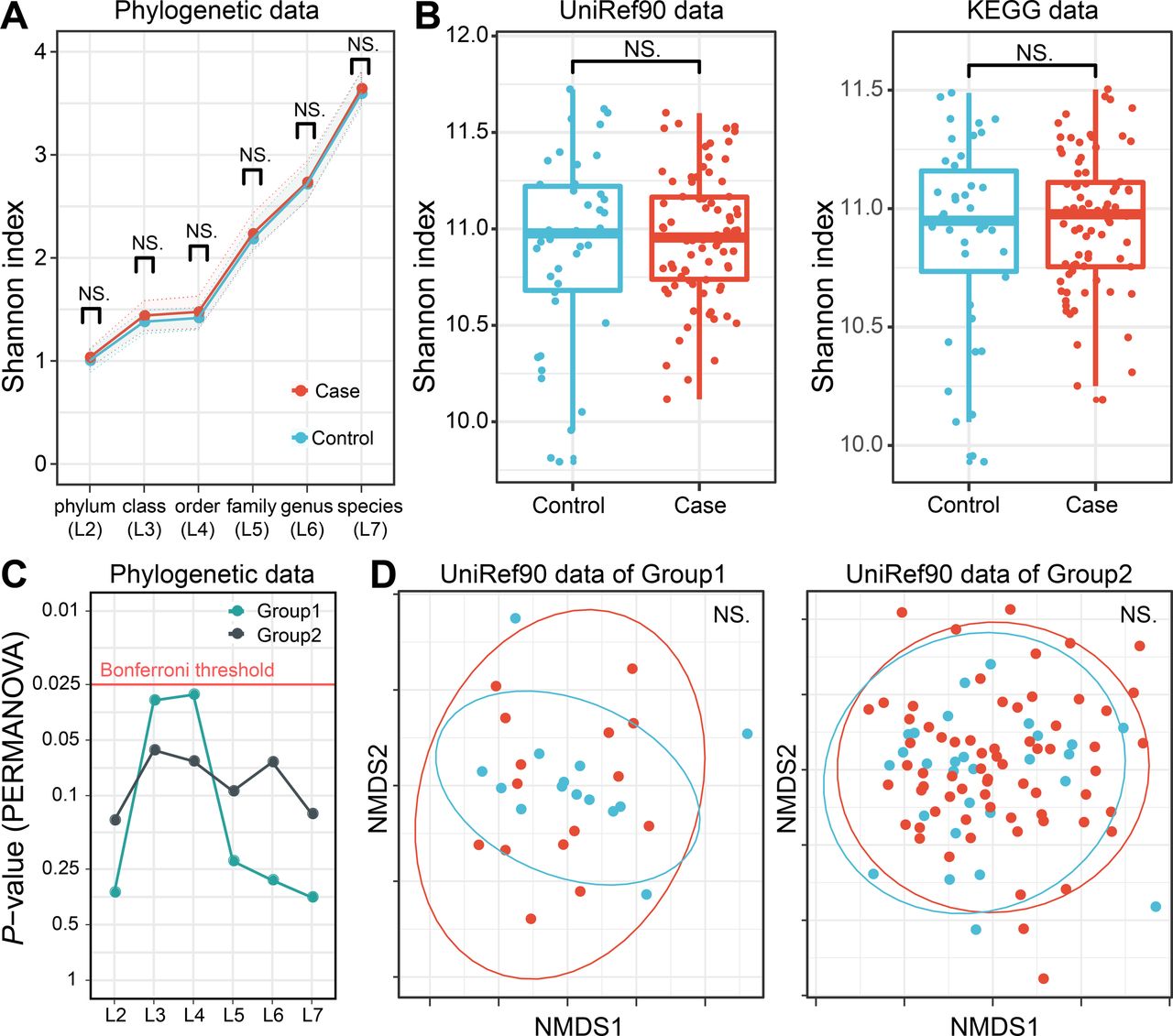
The microbial diversity of the RA gut microbiome is still controversial. We thus evaluated alpha and beta diversity in the phylogenetic data (phylogenetic relative abundance of six levels (L2–L7)) and functional data (gene abundance based on UniRef90 protein and KEGG gene databases). No significant difference was found in any case–control comparisons of alpha diversity based on the Shannon index (p>0.05; figure 4A and B). As for beta diversity, no level of phylogenetic data showed significant case–control differences in either sequencing group (p>0.05; figure 4C). The gene abundance data also did not show significant differences (p>0.05; only UniRef90 data shown in figure 4D). Overall, there was no apparent discrepancy in metagenome diversity between RA cases and controls.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Case–control comparison of microbial diversities in RA. (A) Alpha diversities of the phylogenetic relative abundance data for six levels. Welch’s t-test of Shannon index between RA cases and controls showed no significant difference at any level. (B) Alpha diversities of the gene abundance data of the UniRef90 protein and KEGG gene databases. No significant case–control difference was found. (C) Beta diversities of phylogenetic relative abundance data at six levels. PERMANOVA based on Bray–Curtis dissimilarities found no significant differences among levels for either sequencing group with Bonferroni correction. (D) Beta diversities of the gene abundance of the UniRef90 protein database. No significant case–control difference was found. KEGG, Kyoto Encyclopedia of Genes and Genomes; NMDS, non metric multidimensional scaling; PERMANOVA; permutational multivariate analysis of variance; RA, rheumatoid arthritis.
Discussion
Whole-genome shotgun sequencing of the metagenome has been increasing in importance as more attention is paid to the association between diseases and microbiomes. In our study, we conducted a comprehensive MWAS by using whole-genome shotgun sequencing. We found that the RA gut metagenome of the studied Japanese population has the following interesting characteristics: (1) Multiple species belonging to the Prevotella genus increased in the RA gut metagenome. (2) Non-linear ML methods such as UMAP could be used to successfully deconvolute case–control phylogenetic discrepancies. (3) The abundance of one gene related to redox reaction was decreased in the RA metagenome. (4) Various biological pathways, including those related to metabolism, were enriched in a case–control comparison. (5) A population-specific pathway link between the metagenome (MWAS) and the host genome (GWAS) was identified. (6) No apparent discrepancy in microbial diversity was found between RA cases and controls. Our study greatly exploited the benefits of shotgun sequencing, since it would be challenging to obtain such novel findings using the classical method of 16S rRNA.
Our study is the first to robustly demonstrate that multiple Prevotella spp . other than P. copri increased in the RA gut microbiome by using shotgun sequencing. Recently, Alpizar-Rodriguez et al. reported that Prevotella spp . other than P. copri were enriched in preclinical RA stages, although their assessment was limited to operational taxonomic units assigned at the species level because they used the classical 16S rRNA method and did not conduct statistical analysis.33 Increased levels of microbes of oral origin, including Prevotella, have been reported to be correlated with several diseases.34 35 Atarashi et al. 36 reported that strains of Klebsiella spp . isolated from the salivary microbiota were strong inducers of T helper 1 cells when they colonised the gut.36 In our study, most species of Prevotella showing significant case–control differences were taxa that have been identified in the oral cavity. All species included in the taxa cluster with case–control discrepancies identified by unsupervised ML methods were registered in the expanded Human Oral Microbiome Database.37 The colonisation of the intestine by oral bacteria could be related to the pathogenesis of RA as well as other diseases.
The gene and pathway elements of our MWAS analysis successfully demonstrated the novel functional aspects of the RA gut metagenome. A representative finding was the decrement of a redox reaction-related gene of the genus Bacteroides in RA. It has previously been reported that Prevotella and Bacteroides in the gut in RA showed inverse relationships in terms of their proportional quantity.11 The increase in several species of the genus Prevotella in RA samples further suggests that the balance between these two major taxa in genetic function and composition reflects the disease-specific features of the RA gut metagenome. There could be a possibility that R6FCZ7 and the genus Prevotella were inversely associated via the relationship with the genus Bacteroides, while further functional investigation was required.
The overall association between the metagenome and host genome has mostly been investigated in healthy people38–40; there are few studies focusing on specific diseases. Our MWAS–GWAS interaction analysis demonstrated the population-specific pathway link between the germline genome and metagenome in RA pathology. Further studies investigating biological roles of the detected pathways as well as validating these findings in other independent populations, or in other diseases, are warranted. Furthermore, considering the substantial roles of the human leukocyte antigen (HLA) phenotypes in RA aetiology, the study to investigate the link between HLA phenotypes and metagenome diversity would be also warranted.
To date, discussions on the microbial diversity of the RA gut microbiome have been controversial; some reports have indicated significant differences,10 11 while others have shown no differences.8 41 Recently, a comprehensive analysis of the microbial diversity in many diseases (not including RA) indicated that there were no significant differences in most diseases relative to controls.42 Our results indicate that the same is true for RA, and that the gut microbiome dysbiosis caused by RA would not be sufficient to affect overall microbial diversity. Nevertheless, it would be important how to treat multimapped reads for accurately calculating relative abundances, which is a challenge for further study.
In conclusion, our shotgun sequencing-based comprehensive MWAS in the Japanese RA population revealed a novel link between the gut microbiome, host genome and pathology of RA. In addition to providing novel insights into the RA gut microbiome, our study will provide useful resources for future functional investigations to further elucidate details of the microbiome’s role in RA aetiology.
Acknowledgments
We thank Ms Miho Kaneda for supporting the study.
References
Footnotes
Handling editor Josef S Smolen
Contributors TK and YuO designed the study, conducted the data analysis and wrote the manuscript. YMae, TN, DM, YMat and SN conducted the experiments. YMae, TN, MM, HM, MY, SK, ST, EO, YaO, KK, SH, TH, MN, AO and YS collected the samples. HI, AK, KT and YuO supervised the study.
Funding This research was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (15H05911, 19H01021), the Japan Agency for Medical Research and Development (AMED; 19gm6010001h0004, 19ek0410041h0003, 19ek0109413h0001, 19km0405211h0001), Takeda Science Foundation, Bioinformatics Initiative of Osaka University Graduate School of Medicine, and Grant Program for Next Generation Principal Investigators at Immunology Frontier Research Center (WPI-IFReC), Osaka University.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement The whole-genome shotgun sequencing data are deposited in National Bioscience Database Center (NBDC) Human Database (http://humandbs.biosciencedbc.jp/) with the accession number of hum0197. The data are available upon reasonable request.